
Abstract
HIV-1 protease (PR) represents one of the primary targets for developing antiviral agents for the treatment of HIV-infected patients. To identify novel PR inhibitors, a label-free, high-throughput mass spectrometry (HTMS) assay was developed using the RapidFire platform and applied as an orthogonal assay to confirm hits identified in a fluorescence resonance energy transfer (FRET)–based primary screen of > 1 million compounds. For substrate selection, a panel of peptide substrates derived from natural processing sites for PR was evaluated on the RapidFire platform. As a result, KVSLNFPIL, a new substrate measured to have a ~ 20- and 60-fold improvement in kcat/Km over the frequently used sequences SQNYPIVQ and SQNYPIV, respectively, was identified for the HTMS screen. About 17% of hits from the FRET-based primary screen were confirmed in the HTMS confirmatory assay including all 304 known PR inhibitors in the set, demonstrating that the HTMS assay is effective at triaging false-positives while capturing true hits. Hence, with a sampling rate of ~7 s per well, the RapidFire HTMS assay enables the high-throughput evaluation of peptide substrates and functions as an efficient tool for hits triage in the discovery of novel PR inhibitors.
Introduction
HIV-1 protease (PR), an aspartyl protease, proteolytically cleaves the polyprotein precursors viral gag and gag-pol into functional structural proteins and enzymes. This process is necessary for the maturation of viral particles and infectivity of the nascent virions, an essential step in the HIV-1 replication life cycle. As a result, PR has been identified as one of the primary targets for developing antiviral agents for the treatment of HIV-infected patients. The current standard treatment for HIV infection is highly active anti-retroviral therapy (HAART), which typically combines three or more drugs with complementary mechanisms of action. 1 Protease inhibitors (PIs) are a key component of the HAART regimen and are considered to have a high genetic barrier to the development of drug resistance because of the requirement of the accumulation of several mutations to achieve resistance. A limitation of many currently licensed PIs is the need for coadministration with ritonavir (RTV) or cobicistat to enhance exposure and trough levels. As potent CYP3A4 inhibitors, coadministration with RTV or cobicistat not only substantially increases plasma levels of PIs by blocking their metabolism but also has the potential to interfere with the metabolism of other antiviral agents, which results in an increased propensity for drug-drug interactions as well as metabolic complications. Moreover, chronic treatments with PIs are often associated with several side effects, including lipodistrophy, dyslipidemia, insulin resistance, and risk of cardiovascular disease. Therefore, a critical need remains to develop new PIs that do not require RTV or cobicistat boosting and have reduced incidences of adverse events.
With about 10 PIs approved to treat patients suffering from HIV infection, multiple strategies have been reported to identify inhibitors of PR. Assays monitoring PR activity with high performance liquid chromatography (HPLC) and Western blotting have been developed.2,3 Although effective and PR specific, these assays are not amenable to high-throughput screening (HTS). Enzyme-linked immunosorbant assays have also been reported wherein biotinylated substrates are captured on avidin- or streptavidin-coated plates and antibodies that are specific for either the peptide substrate or the cleavage product are employed for measuring PR activity. 4 However, these assays are technically challenging to automate and suffer from limited substrate concentrations because of the potential for steric hindrance of PR molecules binding to neighboring biotinylated substrates on the plate surface. Moreover, reactions at the solid-liquid interface can be diffusion limited and involve multiple equilibria, which complicates kinetic analysis. Radiometric methods with significantly improved efficiency in measuring PI activities have also been developed but are not ideal for ultra HTS (uHTS) because of the additional cost and safety aspects of working with radioactivity. 5
Multiple light-based strategies amenable to HTS-compatible plate readers have been developed for PR. Early colorimetric endpoint assays were developed by taking advantage of substrate specificity, where PR preferentially cleaves the scissile bond between Phe-Pro, thereby releasing free N-terminal prolyl peptides that are reactive with isatin generating a blue product detectable at 599 nm. 6 In another approach, after proteolytic cleavage of a peptide substrate by PR, the C-terminal product can undergo carbamylation with cyanate followed by condensation with diacetyloxime to generate a color product in the presence of acid, which can be detected at 480 nm. 7 For both of the strategies, involvement of multistep reactions in the detection process makes quantification and reproducibility more challenging for large-scale screening. Continuous assays were also developed to measure the hydrolysis of chromogenic peptide substrates wherein a chromophore, such as p-nitrophenylalanine (NPe), was incorporated in place of the phenylalanine at the P1 or P1′ position in the HIV-1 gag-pol polyprotein, allowing enzyme activity to be monitored spectrophotometrically by ultraviolet (UV) absorbance. 8 However, the elevated Km of the substrate due to these modifications results in low cleavage efficiency of the peptide by PR. Also, a high incidence of compounds interfering with detection via UV absorbance for screening can be disruptive to hit finding in HTS programs.
Alternatively, assays employing fluorogenic substrates were also developed wherein a fluorescent group (o-aminobenoyl) and a chromophoric quencher (NPhe) are placed on either end of the cleavage site. 9 Although the sensitivity was improved relative to UV-based PR assays, the requirement for incorporation of an unnatural amino acid residue NPhe at the P1 or P1′ position had a negative effect on the enzyme’s ability to cleave the substrate. To further improve the assay efficiency, a fluorescence resonance energy transfer (FRET) assay was developed using substrate SQNYPIVQ wherein a donor/acceptor pair of EDANS/DABCYL is conjugated on either end of the peptide, thereby minimizing the impact of the incorporated groups on the affinity of the substrate to PR. 10 This assay format enhances the sensitivity significantly, allowing the substrate to be used at concentrations well below its Km, and therefore becomes a convenient and efficient method for the identification of protease inhibitors.
Although FRET technology is widely used in HTS largely because of the high sensitivity, robustness, and amenability to automation and miniaturization in microtiter plates, it suffers from large numbers of false-positives resulting from compound-based fluorescence interference in screening sets or through compounds binding to fluorescent dyes appended to substrates. 11 Therefore, a nonfluorescent, label-free orthogonal assay is desired to triage and follow-up hits from fluorescent-based HTS. MS presents an attractive method for label-free screening of enzyme targets by directly quantifying native, unmodified substrates and products based on mass-to-charge ratio. However, traditional MS-based assays are limited to throughputs not amenable to HTS paradigms because of the time-consuming liquid chromatography step. The Agilent RapidFire platform addresses this issue by replacing the traditional HPLC unit with a low-volume sample clean-up cartridge combined with a rapid autosampler, enabling analysis of 384-well assay plates at a sampling rate of ~7 s per well. Recently, the RapidFire high-throughput mass spectrometry (HTMS) has been successfully used for the screening of enzymes, such as isocitrate dehydrogenase 1 (IDH1 R132H), 12 kynurenine 3-monooxygenase (KMO), 13 microsomal prostaglandin E synthase-1 (mPGES-1), 14 and histone lysine demethylases, 15 against relatively small collections of compounds. Multiplexing strategies have been used to increase the throughput of the RapidFire to screen libraries of a few hundred thousand compounds,16,17 but it is unlikely that the throughput of the RapidFire will be comparable to many traditional 3456- and 1536-well plate assay formats, making routine screening of libraries of >1 million compounds challenging. However, the RapidFire system is compatible with throughput requirements for HTS hit triage or follow-up. To date, an MS-based assay has not been reported for HTS of PR. Herein, we report a strategy to identify PR inhibitors wherein a screen of a library of >1 million compounds in a FRET-based µHTS assay is coupled with an MS-based confirmatory assay for compound progression. The details of the HTMS assay development including optimization and evaluation of peptide substrates on the RapidFire platform are described.
Materials and Methods
Reagents and Materials
The SensoLyte 520 HIV-1 protease FRET assay kit was obtained from AnaSpec Inc. (Cat No. 71147; Fremont, CA). The PR peptide substrate
HIV-1 Protease FRET Primary and Does-Response Screens
The primary screen of >1 million compounds was performed in a FRET assay at a compound screening concentration of 10 µM in 3456-well plate format at a final volume of 1 µL per well. Using an ATS100 acoustic liquid dispenser (EDC Biosystems, Fremont, CA), 5 nL of a 2 mM stock of compound diluted in DMSO was preplated into untreated, black 3456-well assay plates. Then, 0.5 µL of 40 nM PR in assay buffer (50 mM NaOAc pH 5.5, 100 mM NaCl, 0.01 % [w/v] BSA, 0.0015 % Triton X-100) was dispensed with a BioRaptr flying reagent dispenser (Beckman Coulter, Pasadena, CA) to the wells. Plates were incubated at room temperature for 15 min, and then 0.5 µL of 2 µM SensoLyte 520 HIV-1 protease substrate (Km = 7 µM) was dispensed into all wells to initiate the reaction. After incubating for 90 min at room temperature, plates were transferred immediately to a ViewLux reader (Perkin Elmer, Waltham, MA), and the fluorescence intensity was measured over 20 s (excitation filter 480/20 nm; emission filter 540/25 nm).
For the dose-response screen, 2 µL of 25 nM PR in assay buffer was dispensed with a BioRaptr flying reagent dispenser to an untreated, black 1536-well plate. Using a Janus pintool (PerkinElmer, Waltham, MA), 30 nL of compound diluted in DMSO was transferred followed by a 15 min incubation at room temperature. Reactions were initiated with the addition of 0.5 µL of SensoLyte 520 HIV-1 protease substrate (1:20 dilution in assay buffer, 5 µM) into all wells. Plates were incubated at room temperature for 90 min and then immediately read on a ViewLux reader (excitation filter 480/20 nm; emission filter 540/25 nm). Compounds were titrated in duplicate using an eight-point, threefold dilution series starting from a top concentration of 100 µM. Pepstatin A, a known HIV protease inhibitor, was used as a positive control in both primary and dose-response screens at its IC50 value of 263 nM.
HIV-1 Protease HTMS Confirmation Assay
The PR HTMS confirmation assay was performed in 384-well plate format on the Agilent RapidFire RF300 system (Agilent, Wakefield, MA) coupled to an API-4000 mass spectrometer (AB Sciex, Framingham, MA). Fifteen microliters of PR in assay buffer (50 mM NaOAc, pH 5.5, 100 mM NaCl, 0.0015% Triton X-100, 0.01% [w/v] BSA) was added to wells using a BioRaptr dispenser. Then, 0.1 µL of each compound was transferred with a Janus pintool to each well and incubated at room temperature for 15 min. Reactions were initiated with the addition of 5 µL peptide substrate in assay buffer followed by a 60 min incubation at room temperature. The final screening conditions are 1.6 nM PR, 60 µM peptide substrate
RapidFire Mass Spectrometry
The quenched 384-well assay plates were spun at 1300g for 5 min at room temperature and then transferred onto the Agilent RapidFire RF300 HTS platform. Ten microliters of sample from each well was aspirated and loaded onto a micro-scale C4 solid-phase extraction (SPE) cartridge, where nonvolatile assay components such as salts, buffers, and detergents that are incompatible with MS were removed by washing with solvent A (water containing 0.1% [v/v] formic acid and 0.01% [v/v] TFA) at a 1.5 mL/min flow rate in a 3 s wash cycle (equivalent to three-column volumes). The purified analytes of interest retained were then eluted off the cartridge using solvent B (80% acetonitrile containing 0.1% [v/v] formic acid and 0.01% [v/v] TFA) at a flow rate of 1.25 mL/min, directly into a AB Sciex API-4000 triple-quadrupole MS for analysis. Reequilibration of the SPE cartridge was achieved with a 0.5 s wash with solvent A at a flow rate of 1.5 mL/min. The aspirator tip was washed using water and acetonitrile between sample aspirations (each for 500 ms) to minimize carryover contamination. MS was carried out using electrospray ionization in the positive ion mode (with the exception of substrate
Data Analysis
The MS chromatogram area under the daughter ion peaks of each component (area under the curve [AUC]) was integrated using the RapidFire integrator software. The data for each well were analyzed by either monitoring substrate conversion P/(P+S), where P = MS AUC for the product and S = MS AUC for the substrate or normalization of data to an internal standard (P/IS), where IS = MS AUC for the internal standard. The percentage of inhibition (%INH) was calculated with respect to the high and low controls by the following formula: %INH = [1 − (Normalized Compound Response − Normalized High Control Response)/(Normalized Low Control Response − Normalized High Control Response)] × 100%. Offline data for IC50 determination of control compounds were analyzed by Prism 5 (GraphPad, San Diego, CA) using nonlinear regression curve-fitting algorithms (variable slopes).
Results and Discussion
HIV-1 Protease Primary Screen in the FRET Assay
The FRET assay for the PR primary screen in 3456-well plate format was developed based on the SensoLyte 520 HIV-1 protease assay kit protocol with some modifications, in which PR activity is quantified using a FRET peptide substrate incorporated with a HiLyte Fluor 488 (fluorophore) and QXL 520 (quencher). The sequence of this FRET peptide is derived from the native p17/p24 cleavage site on Prgag for PR. Initially in the intact FRET peptide, the fluorescence of HiLyte Fluor 488 is quenched by QXL 520. Upon substrate cleavage by PR, the fluorescence is recovered and can be continuously monitored at excitation/emission = 490 nm/520 nm. With a longer emission wavelength, this FRET peptide is expected to have less interference from the autofluorescence of test compounds. During assay development, PR was titrated in concentrations ranging from 1.25 nM to 40 nM, and the reaction was monitored for 110 min. The fluorescent response to PR concentration was linear with respect to time, resulting in the selection of 20 nM PR and a 90 min reaction time for the screen due to a signal to background (S/B) of approximately 18:1 ( Fig. 1A ). Various additives to the assay buffer were investigated for the reduction of nonspecific binding of compounds to PR in the assay. It was found that the presence of 0.01% (w/v) BSA in the assay buffer reduced the screening hit rate from 2.7% to 0.9% based on the mean +3-sigma cutoff for the assay. To benchmark the assay, Pepstatin A, a known PR inhibitor, was titrated under the optimized assay conditions and determined to have an IC50 value of 263 nM (Ki = 230 nM), which is comparable to previously published values for this compound ( Fig. 1B ). 19 The incorporation of 0.01 % (w/v) BSA in the assay buffer was found to have no measurable effect on the enzyme activity or inhibition of the enzyme by Pepstatin A.
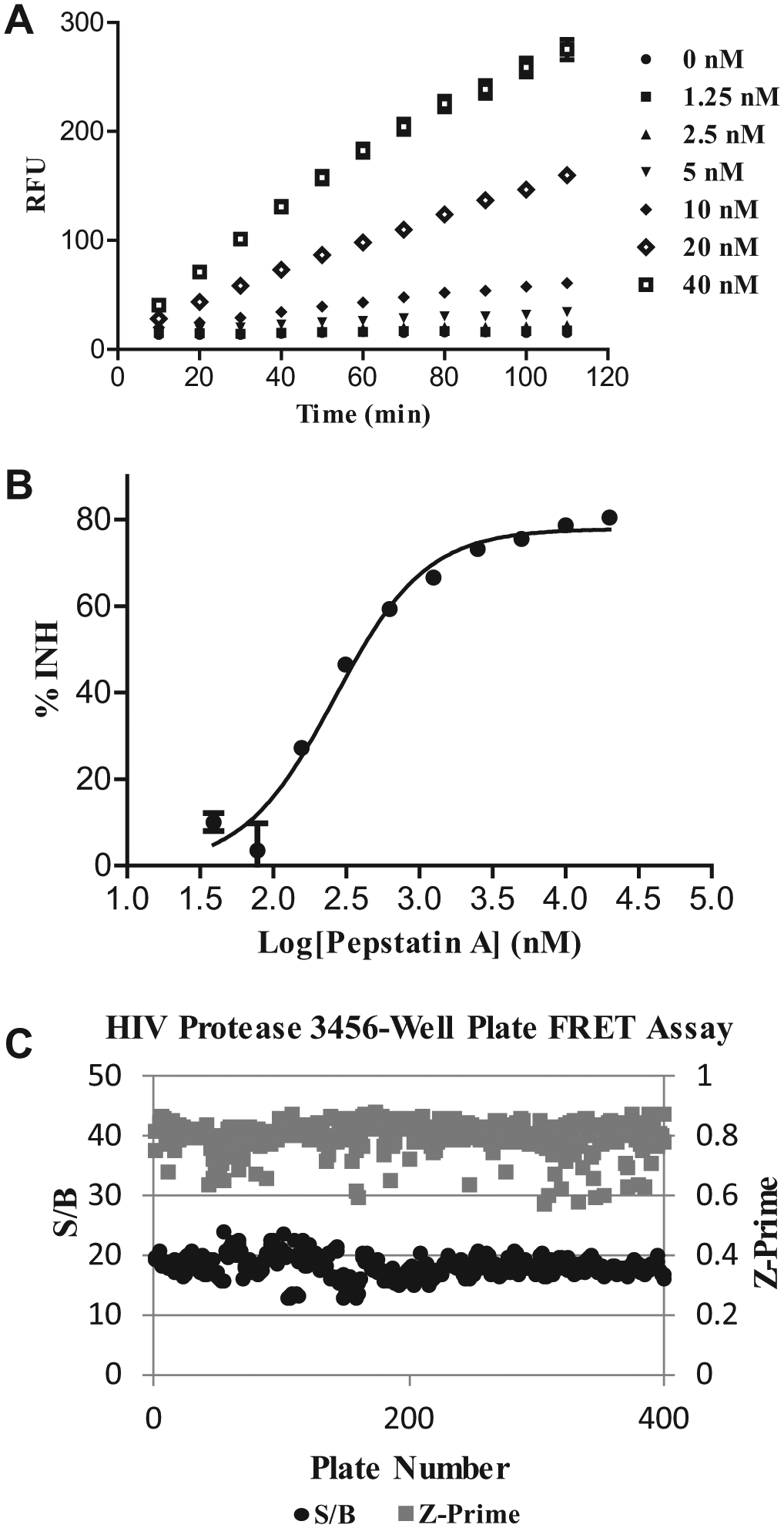
HIV-1 protease fluorescence resonance energy transfer (FRET) assay development. (
Prior to the screen, the robustness of the FRET assay was examined by screening of a subset of 6000 compounds at a single concentration. The assay was robust in HTS, with a mean Z′ of 0.8. The primary screen of a >1 million compound library was completed within 2 wk in the 3456-well plate FRET assay at a screening concentration of 10 µM of each compound per well. Pepstatin A was spiked in the control region of each assay plate as a screening quality control during the HTS campaign. For the duration of the screen, Z′ values were measured within the range of 0.6 to 0.85 with a mean 18-fold assay window ( Fig. 1C ). Traditionally, hits were selected from the primary uHTS screen based on the statistical cutoff of the mean +3-sigma for the assay. However, systematic plate effects during the operation of the screen can bias the hit selection. Therefore, we adopted a strategy to minimize the effects of systematic plate effects on hit selection by selecting hits based on the four different scoring approaches including % activity, % activity adjusted based on the B-score algorithm, B-score, and Z-score computed for each well of each plate. 20 The top ranking compounds (top 1%) were selected for each scoring approach, and the union of all four hit sets was taken as the final hit list, which resulted in the selection of 24,032 compounds for confirmation.
HTMS Assay Development
The first step in the development of the PR HTMS confirmatory assay required the selection of an optimal peptide substrate. Many peptide substrate sequences derived from natural processing sites of PR have been reported, although Km values for the majority of the unlabeled peptides are reported in the millimolar range.10,21 Appending the substrates with fluorophores enhanced binding and reduced Km values to the high micromolar range. 10 However, fluorescent labels can generate false-positives in assays because of compounds’ interacting directly with the hydrophobic fluorescent dyes.22,23 An unmodified peptide substrate with improved Km and cleavage efficiency properties is desired for the HTMS assay.
Several studies have focused on optimizing the Km and catalytic efficiency of PR substrates. Although certain common features were observed, comparison of cleavage site sequences indicates that PR substrates lack a consensus sequence resulting in a broad substrate specificity.24,25 The analysis of crystal structures of substrate complexes also suggests that substrate shape rather than a particular amino acid sequence determines Km and catalytic efficiency. 26 This is largely due to the preference of the enzyme for a residue at a particular position in the substrate being dependent on the neighboring residues. Therefore, random screening systems including phage display, traditional HPLC/MS, and fluorescence assays have been used for the systematic evaluation and optimization of peptide substrates for proteases.25,27,28 With the ability to quantify native peptide substrates and their corresponding products at a sampling rate of ~7 s per sample, the RapidFire HTMS provides an ideal platform for the evaluation of peptide substrates in a high-throughput manner. In this case, the evaluation of 10 unlabeled peptide substrates was completed within 2 wk on the RapidFire platform. Among the panel of 10 substrates evaluated, 6 were selected from literature reports of substrates used in various PR assays including SQNYPIV and SQNYPIVQ, the most commonly used sequences representing the naturally occurring cleavage site between the matrix and capsid proteins. 10 Four additional new peptide sequences were designed to improve Km and cleavage efficiency properties as described below.
It was reported that the hydrophobic residues PIV are the most favorable at the P1′-P3′ position of PR substrates, and the shortest peptide sequence that still maintains good cleavage efficiency by PR consists of five amino acids on the P side of the cleavage site and three amino acids on the P′ side. 21 Substrate optimization was therefore focused on peptide sequences with five amino acids on the P side and the PIV sequence on the P′ side. Exploration of the susceptibility of natural cleavage sites in HIV-1 Gag and Gag-Pol polyproteins to PR suggested that the sequence of VSFNFPQV exhibits high hydrolytic efficiency by PR. 29 When various sequences with changes to amino acids between the P2 and the P4 position were tested, the sequence with the highest level of cleavage efficiency contained Leu at the P3 position. 30 Based on this result, VSLNF was selected as the sequence for the position P5-P1. Combined with the residue PIV, the sequence of VSLNFPIV was selected as a substrate for evaluation in the HTMS assay. In addition, previous reports have identified Leu as the optimal residue at the P3′ position. 24 Therefore, the sequence of VSLNFPIL was also synthesized for evaluation in the assay. To improve solubility and the ionization potential of the substrates by electrospray in the HTMS assay, peptides KVSLNFPIL and KVSLNFPIV were designed with the addition of a lysine to the N-terminus of the peptide sequences. Interestingly, the addition of the lysine in both peptides also enhances the catalytic efficiency by PR.
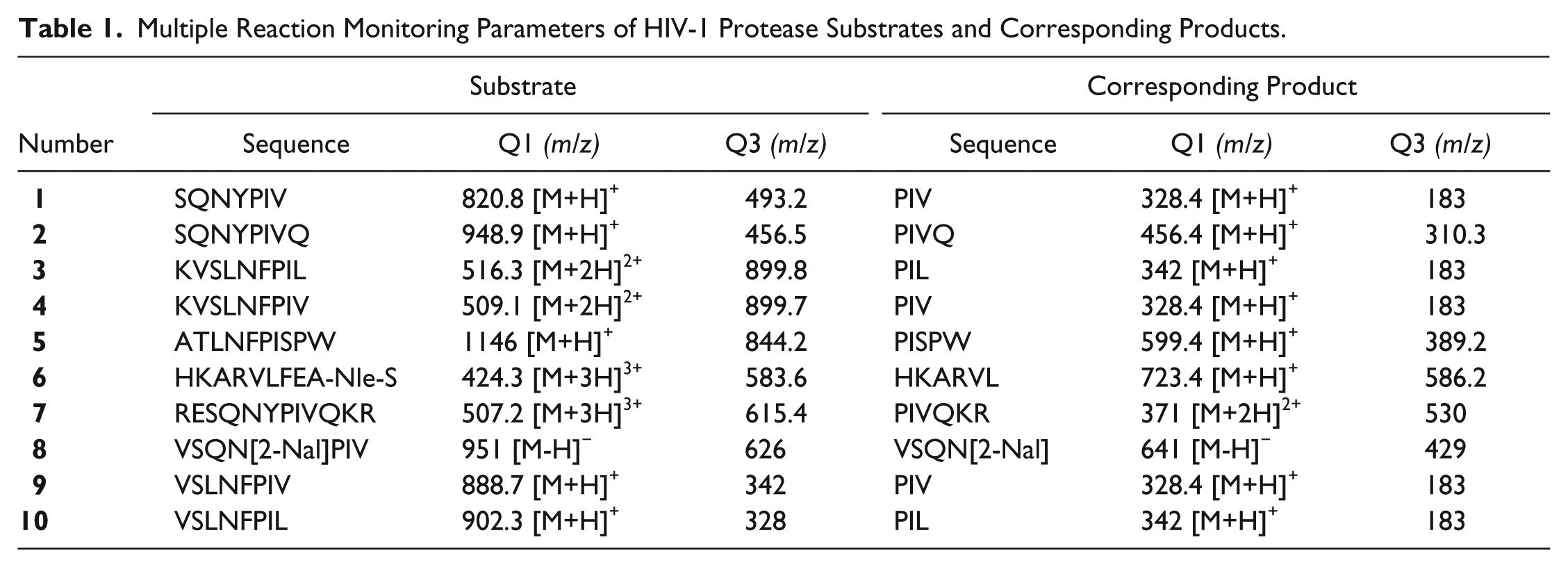
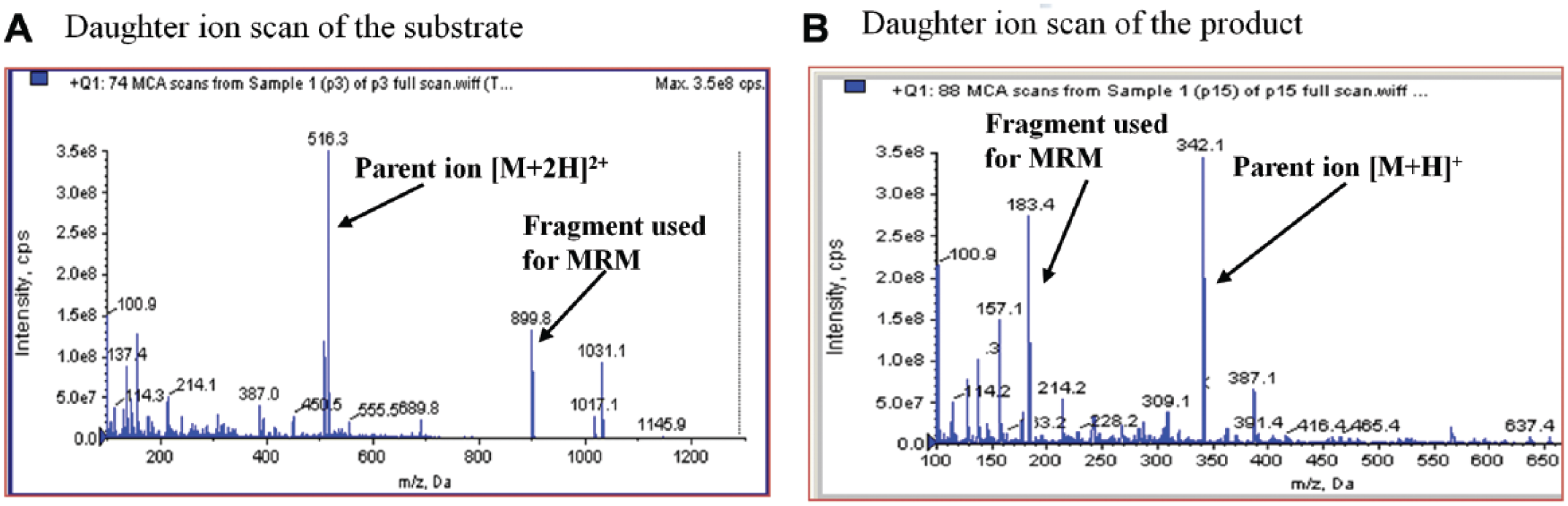
To evaluate the peptide substrates in the HTMS assay, we first identified the prominent mass ions from each substrate and its product that can be used in the HTMS assay to quantify PR activity (
Table 1
). Each peptide was dissolved individually in acetonitrile/water (1:1) at a concentration of 0.5 µM and subjected to analysis using an API-4000 triple-quadrupole electrospray MS. MS parameters including ionization mode, DP, and CE were optimized for each substrate and its product (data not shown here). The technique of MRM was used as a quantitative detection method to selectively discriminate desired parent and daughter ion pairs from contaminating mass species, providing the selectivity and sensitivity required for quantifying the analytes of interest. A representative full ion scan acquired in positive mode on the mass spectrometer is shown in
Figure 2
for peptide substrate
Multiple Reaction Monitoring Parameters of HIV-1 Protease Substrates and Corresponding Products.
Optimization of substrate
To select the optimal substrate for further HTMS assay development, a time course experiment was performed at room temperature for each substrate at a concentration of 1 µM with 5 nM PR. The reactions were subsequently quenched at the reported time point with 20 µL of 2% formic acid in water and subjected to analysis using the RapidFire platform. Each injection of the sample was followed by an injection of the assay buffer to ensure no carryover from sample to sample. Product formation followed linear kinetics with respect to time, and the percent conversion of each substrate was determined (
Fig. 3
). Low percentage conversion of the substrate was observed for substrates
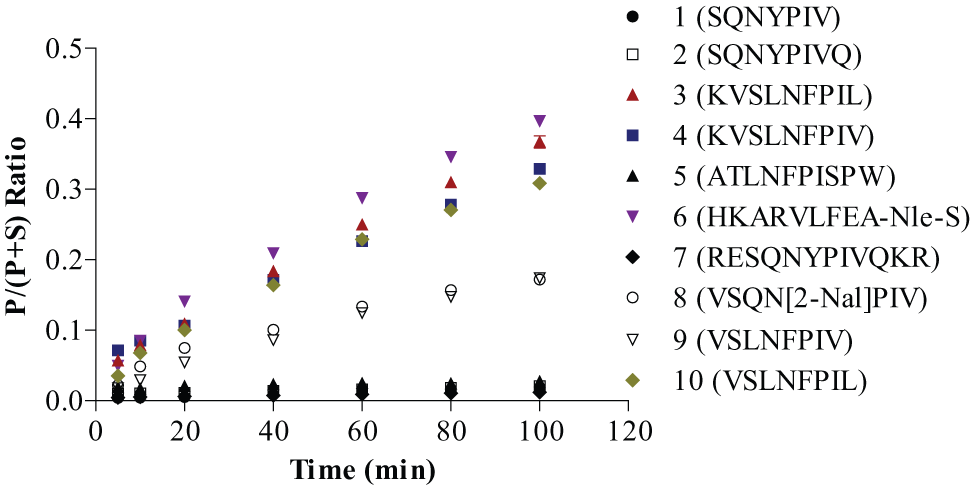
Comparison of hydrolysis rate of HIV-1 protease substrates. Reactions were carried out at 5 nM HIV-1 protease and 1 µM substrate individually in the assay buffer and quenched by 2% formic acid at the individual time point. Each point represents mean ± SD for n = 3 replicates.
Kinetic parameters were measured for the four selected peptide substrates. The Km of the enzyme for substrate
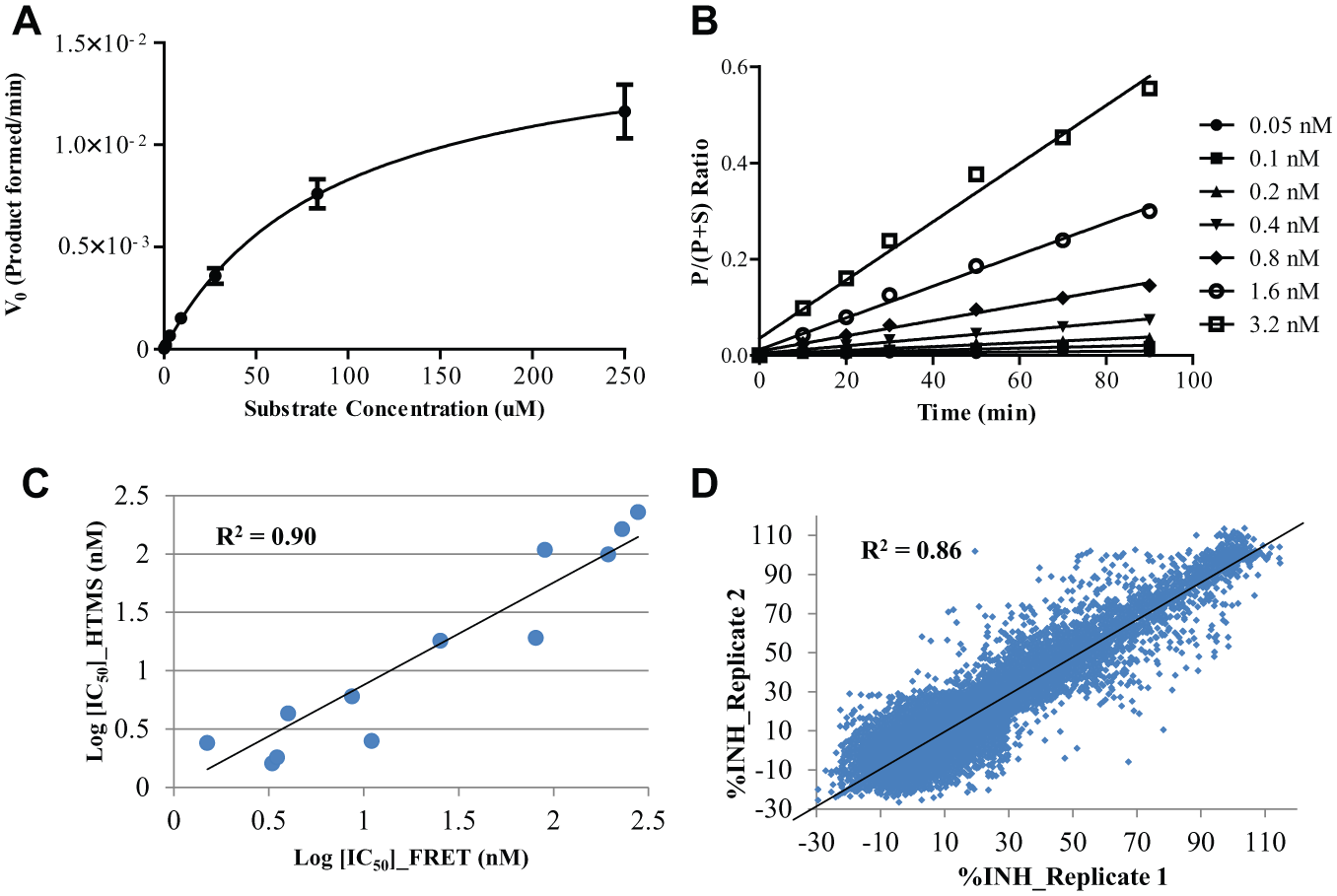
Description of HIV-1 protease high-throughput mass spectrometry (HTMS) assay with substrate
Enzyme Kinetics of Selected HIV-1 Protease Substrates.
To determine the optimal enzyme concentration for the HTMS assay, PR was titrated at various concentrations (0.05 nM to 3.2 nM) at a fixed concentration of substrate
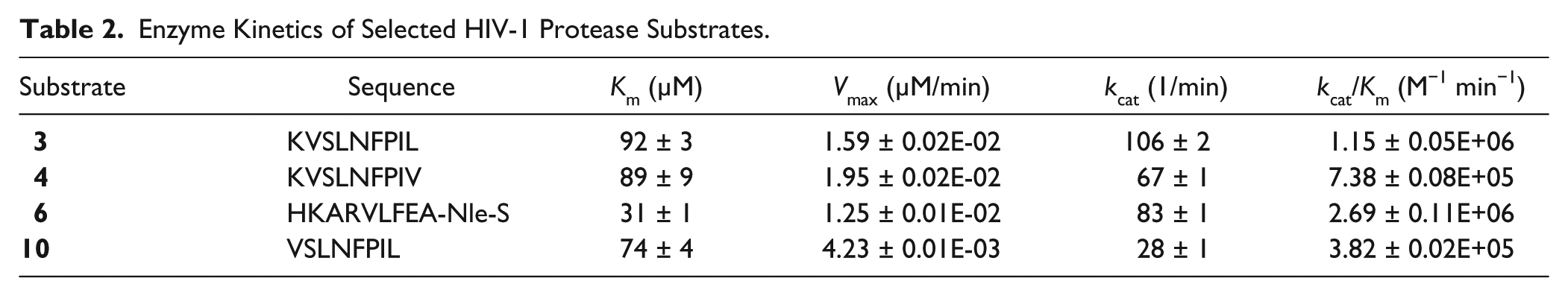
Other variables optimized during assay development included the comparison of the quench solutions and mobile phases, as well as the implementation of an internal standard. Quench solutions of methanol, acetonitrile, and 2% (v/v) formic acid in water were evaluated in the HTMS assay. A reaction quench of 2% (v/v) formic acid in water was selected for the screen because it completely stopped the reaction and consistently resulted in higher signal of the peptide product and lower data variation. A stable isotope-labeled variant of the product ([P-U6]IL) was premixed with the reaction quench solution at a final assay concentration of 2 µM and used as an internal standard for the HTMS screen. The employment of this internal standard, which was co-eluted and detected along with the product, allowed for normalization of the assay data to minimize effects from the process and detection-related variability. Studies were also performed to identify the optimal mobile phase for elution of analytes from the SPE cartridge. Slightly tailed peaks for both the substrate and the product were observed in the MS chromatogram when eluting with 100% acetonitrile containing 0.1% formic acid and 0.01% trifluoroacetic acid, while elution with 80% acetonitrile containing 0.1% formic acid and 0.01% trifluoroacetic acid resulted in well-resolved peaks with no evidence of carryover detected from sample to sample. DMSO plates were used to measure full plate statistics for the HTMS assay, which yielded an average S/B = 8 and a Z′ of ~0.8. Finally, a high correlation was observed (R2 = 0.9, the slope of the best-fit line = 0.94) for the IC50 values for a panel of 12 known PR inhibitors titrated in both the FRET and the HTMS assays ( Fig. 4C ).
Results of the HIV-1 Protease Inhibitor Screen
The primary screen of >1 million compounds in a 3456-well plate FRET assay against PR resulted in 24,032 putative hits. As expected, the confirmation screen of a subset of 2560 primary hits run in triplicates in the same FRET assay yielded a high confirmation rate of ~80% based on a cutoff of 30% inhibition, which corresponds to the mean +3-sigma for the assay. For compound progression, a more efficient orthogonal confirmation assay must be incorporated to remove false-positives and progress true actives. MS, a label-free technology that is capable of directly quantifying native, unmodified substrates and products based on the mass-to-charge ratio, has proven to be an attractive method for reducing fluorescent-related false-positives. The HTS hits selected from the primary screen were therefore confirmed using the HTMS assay in duplicate in 384-well plate format with high reproducibility (R2 = 0.8, the slope of the best-fit line = 0.93; Fig. 4D ). The HTMS assay plates consistently performed well, with an average S/B = ~8 and Z′ = ~0.7. From the HTMS confirmation screen, hits were defined as compounds demonstrating >30% inhibition (mean +3-sigma cutoff) for the assay in either replicate to avoid missing true hits in which one of the replicates are low due to a robotic error or a missed injection. Based on these criteria, only 4179 compounds (corresponding to a ~17% confirmation rate) were confirmed as hits to progress to the dose-response assays. After removal of 304 known PR inhibitors, 3875 compounds were progressed and titrated in duplicate in the same FRET assay as the primary assay but in 1536-well plate format. From this set, 202 compounds were calculated to have submicromolar potency (IC50 <1 µM) against PR. An additional 529 compounds were calculated to have IC50 values between 1 µM and 10 µM. Several structural scaffolds were identified that demonstrated tractable SAR by medicinal chemistry (data not shown).
Herein, we have described a strategy to screen >1 million compounds using a light-based FRET assay followed by a label-free, MS-based assay for hit triage. This approach minimizes the artifacts from fluorescence interference and progresses true hits into downstream follow-up. As in this case, about 17% of hits were confirmed in the HTMS assay in contrast to an estimated 80% confirmation rate in the FRET assay. The relatively large difference of confirmation rates between the HTMS assay and the FRET assay could potentially result from many causes including fluorescent interference, substrate-compound interactions, and substrate-specific inhibition. The label-free nature of detection in HTMS assays eliminates the risk of fluorescent interference commonly seen in FRET assays. However, substrate-compound interactions and substrate-specific inhibition must also be considered due to the structural difference between sequences of the peptide substrates in the two assays. Importantly, all 304 known PR inhibitors in the set were identified in the HTMS assay, demonstrating that this approach effectively reduces false-positives while capturing true hits. Substrate selection is also an important aspect of the HTMS assay development. A panel of unlabeled peptide substrates was evaluated on the RapidFire platform within 2 wk. A new peptide substrate KVSLNFPIL, which is about 20- to 60-fold more catalytically efficient than the previously reported substrates SQNYPIVQ and SQNYPIV, was identified for the HTMS screen. Compared with fluorescent-labeled substrates, the use of unlabeled substrates not only streamlines assay development by eliminating the expense and additional time required for the generation and validation of labeled substrates but also reduces potential false-positives due to compounds interacting directly with hydrophobic fluorescent dyes. Hence, with a sampling rate at ~7 s per well, RapidFire HTMS provides an ideal platform for evaluation and optimization of unlabeled peptide substrates in a high-throughput manner and the enrichment of true actives from uHTS hit lists.
Footnotes
Acknowledgements
We thank Dr. Andy Liaw and Anthony Kreamer for selecting uHTS primary hits and for bioinformatics support; Edward Hudak and Alex Wolicki for compound management support; Dr. Sujata Sharma, Dr. Keith Rickert, and Dawn Hall for supplying HIV-1 protease. Also, we would like to thank Dr. William LaMarr, Dr. Patty Sun, Dr. Kari Schlicht, and Dr. Maxine Jonas of Agilent Technologies for help with the RapidFire instrument setup and technical support.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.