
Abstract
G-protein-coupled receptors (GPCRs) are crucial cell surface receptors that transmit signals from a wide range of extracellular ligands. Indeed, 40% to 50% of all marketed drugs are thought to modulate GPCR activity, making them the major class of targets in the drug discovery process. Binding assays are widely used to identify high-affinity, selective, and potent GPCR drugs. In this field, the use of radiolabeled ligands has remained so far the gold-standard method. Here the authors report a less hazardous alternative for high-throughput screening (HTS) applications by the setup of a nonradioactive fluorescence-based technology named Tag-lite®. Selective binding of various fluorescent ligands, either peptidic or not, covering a large panel of GPCRs from different classes is illustrated, particularly for chemokine (CXCR4), opioid (δ, µ, and κ), and cholecystokinin (CCK1 and CCK2) receptors. Affinity constants of well-known pharmacological agents of numerous GPCRs are in line with values published in the literature. The authors clearly demonstrate that the Tag-lite binding assay format can be successfully and reproducibly applied by using different cellular materials such as transient or stable recombinant cells lines expressing SNAP-tagged GPCR. Such fluorescent-based binding assays can be performed with adherent cells or cells in suspension, in 96- or 384-well plates. Altogether, this new technology offers great advantages in terms of flexibility, rapidity, and user-friendliness; allows easy miniaturization; and makes it completely suitable for HTS applications.
Introduction
G-P
Fluorescence-based techniques therefore have been investigated to propose alternatives to radioactivity in the development of binding assays. In particular, the DELFIA® technology, based on the use of lanthanide chelates as fluorescent probes, was developed in the past decade. 4 Taking advantage of the millisecond lifetimes of the lanthanide complexes by gating the emission output greatly reduces the background fluorescence generated in the sample, allowing a high assay sensitivity. However, washing steps remain mandatory to set up DELFIA binding assays and thus limit their miniaturization.
Fluorescence polarization (FP) has become increasingly popular due to its ease of operation and its miniaturization possibilities. Since FP does not require any receptor tagging, binding assays can be carried out on wild-type receptors or on endogenous receptors if well expressed. However, it has several drawbacks such as (1) fluorescent ligand mass should be below 5 kDa and have high affinity (below 5 nM), 2 (2) fluorescence background from chemical compounds or biological material can affect the accuracy of FP, and (3) the hydrophobic nature of some small fluorescent ligands can induce significant nonspecific binding to cell membranes, resulting in a small output signal.
Fluorescence correlation spectroscopy (FCS) is a promising technique based on fluorescently labeled ligands that can be performed on living cells without any washing steps. Like FP, FCS assays can be carried out on wild-type receptors or on endogenous receptors. However, the complexity of the instrumentation and its relatively low throughput limit its HTS capabilities.
More recently, several fluorescence resonance energy transfer (FRET)–based GPCR binding assays have been described. 5 One well-known illustration is based on the combination of a green fluorescent protein (GFP)–fused GPCR and a near infrared fluorescent ligand that allows the identification of new compounds inhibiting the interaction of CXCR4 with its natural ligand. 6 However, the assay displayed both limited signal to background (S/B ratio) and miniaturization capabilities. Another assay format proposed a displacement assay for β2-adrenergic receptor based on quenching resonance energy transfer with lanthanide chelates, 7 but this technique is unlikely to be used for many GPCRs due to the unpredictable quenching efficiency of the bound chelate. Time-resolved FRET assays based on the noncovalent labeling of the receptors with anti-tag antibodies have already been developed. 8 These assays exhibit a good sensitivity. However, they do not allow the determination of the affinity of the competitor since the assay equilibrium depends both on antibodies and tracer binding kinetics.
To overcome this limitation, we present a new Tag-lite® ligand-binding assay format on living cells that turns out to be widely applicable to a whole range of GPCRs. These assays are based on the combination of a Homogeneous Time-Resolved Fluorescence (HTRF®) detection method with a covalent labeling technology called SNAP-tag®, previously developed by Keppler and coworkers. 9
HTRF is a homogeneous technology that combines a FRET process with a time-resolved fluorescence detection used to probe biomolecular interactions. 10 This combination, named TR-FRET, is possible through the use of long lifetime fluorescent FRET donors such as europium or terbium cryptates. Terbium cryptate (Lumi4®-Tb) is of particular interest, thanks to the negligible emission around 520 nm and 665 nm and its energetic compatibility with both green and red fluorescent dyes, which can therefore be used as acceptors. This flexibility in the choice of acceptor broadens the development possibilities of appropriate ligands for a given GPCR that remain potent once modified, by keeping its binding affinity and pharmacological properties. When both acceptors behaved the same, our focus was on the development of red fluorescent ligands that display the least possible compound interference.
To set up a Tag-lite binding assay of interest, the targeted GPCR, fused at its extracellular N-terminal part with the SNAP-tag moiety, is expressed at the surface of living cells. The addition of nonpermeant SNAP-tag substrates derivatized with the highly emissive Lumi4-Tb (SNAP-Lumi4-Tb) allows the labeling of SNAP-tag-fused GPCR with the FRET donor of interest through a covalent bond ( Fig. 1A ). A previous study showed that cell surface GPCRs fused with the SNAP-tag can be labeled with 100% efficacy and that the 23-kDa SNAP-tag 9 moiety linked to the GPCR does not alter its expression and its functional behavior. 11 Upon binding of a fluorescent ligand bearing either a green or a red acceptor on the donor-labeled SNAP-tag/GPCR fusion protein, an HTRF signal from the sensitized acceptor can be detected since the energy transfer can occur only when the donor and the acceptor are in close proximity ( Fig. 1B ). Inhibition of the HTRF signal after competitive binding with a nonlabeled compound can therefore be used to identify and characterize new GPCR ligands from a compound library. The distance dependence of the HTRF signal ensures a high specificity of the assay for the GPCR of interest.
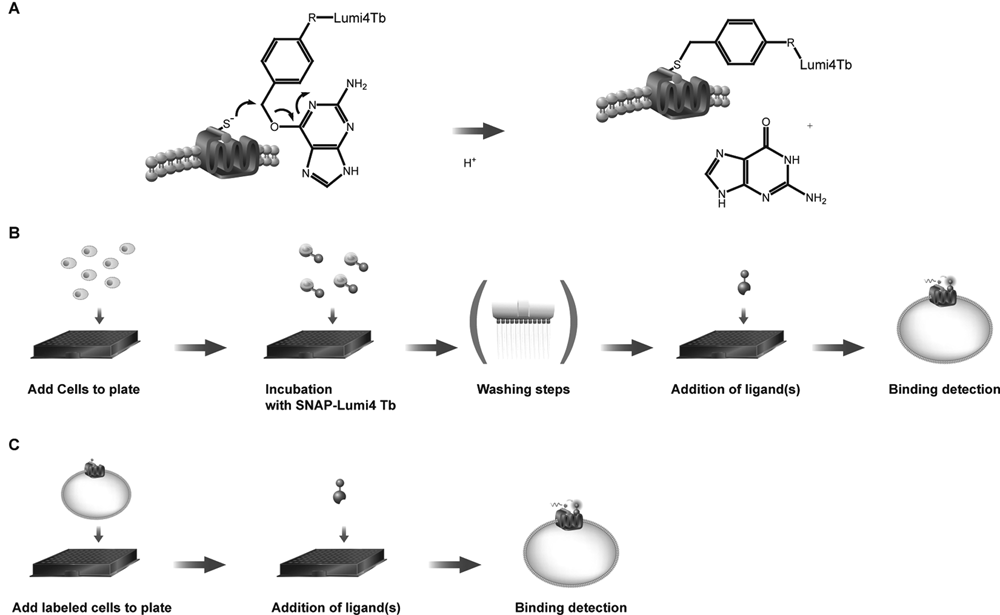
Tag-lite binding assay principles. (
Ligand-binding assays on adherent cells have been successfully developed using this concept for a whole range of GPCRs using both peptidic and nonpeptidic fluorescent ligands. Using either transiently transfected cells or stable cell lines, competitive binding assays with various agonists and antagonists of these receptors result in an accurate pharmacological characterization of these compounds, in complete correlation with the data available in the literature. In particular, we highlight here the development of assays for the cholecystokinin (CCK1 and CCK2) receptors, the CXCR4 chemokine receptor, and the 3 subtypes of opioid receptors (µ, δ, and κ).
Cholecystokinin peptides play key regulatory functions in the digestive tract and nervous system through their binding on either CCK1 or CCK2 receptors. CCK1 predominates in the periphery, whereas CCK2 represents the predominant receptor population in the central nervous system. 12 It is implicated in psychiatric disorders, including anxiety, panic attacks, and pain perception. To our knowledge, binding assays based on TR-FRET/HTRF were not reported for these receptors. To achieve their development, a nonselective fluorescent agonist was synthesized. By using this fluorescent agonist, binding assays both for CCK1 and CCK2 receptors were established and showed good correlation with existing assays and data.
CXCR4 is of particular interest for drug development due to its implication in many diseases such as cancer and HIV infection. The optimization of new tools for the screening of chemokine receptors has recently resulted in the development of a wide variety of assay formats, including fluorescent ligands for binding assays, flow cytometry, 13 or FRET assay, 6 but also genetic fusion of GFP to SDF-1α 14 and assays using a bioluminescent SDF-1α fusion protein. 15 Using a fluorescent derivative of SDF-1α/CXCL12, the natural CXCR4 agonist, we established a Tag-lite CXCR4 binding assay. Because SDF-1α/CXCL12 can also bind cell surface proteoglycans, 16 the HTRF signal resulting from the binding of the fluorescent SDF-1α to the labeled SNAP-tag-CXCR4 is of great advantage to avoid nonspecific signal arising from this interaction with glycans. Therefore, the Tag-lite binding assay presented in this article shows high specificity and is well suited to characterize new CXCR4 ligands.
For the opioid receptors, well known for their implication in pain, a heterogeneous lanthanide-based ligand-binding assay was previously described for the δ-opioid receptor using fluorescent enkephalin peptide as the tracer. 4 However, since this fluorescent ligand is highly selective for the δ subtype, it cannot be used to probe other opioid receptor subtypes. To overcome this limitation, we designed a fluorescent derivative of a nonpeptidic compound, naltrexone. This compound is a nonselective opioid antagonist displaying a high affinity for all the opioid receptor subtypes (µ, δ, and κ). Using this single fluorescent ligand, we have been able to set up Tag-lite binding assays suitable for the study of the 3 opioid receptor subtypes.
These assays, first developed on adherent cells, were also adapted for the binding to be carried out using frozen cells in suspension, labeled in advance with SNAP-Lumi4-Tb. This mix-and-measure format is a straightforward binding assay and is particularly suitable in an HTS environment by providing a miniaturized format and an attractive alternative method to replace radioactive assays.
Materials And Methods
Reagents
The Tag-lite labeling medium was from Cisbio Bioassays (Bagnols-sur-Cèze, France; ref. LABMED). The 96-well plates (ref. 655086) as well as the 384-well small volume plates (ref. 784075 and 784076) were purchased from Greiner Bio-One (Monroe, NC).
O6-Benzylguanine-Lumi4-Tb was synthesized by Cisbio Bioassays and is commercialized as SNAP-Lumi4-Tb (ref. SSNPTBE).
A naltrexone derivative labeled with a red fluorescent probe (red-naltrexone; Cisbio Bioassays, ref. L0005RED) was used for the µ-, δ-, and κ-opioid receptor binding assays. For the CXCR4 receptor, a red fluorescent analogue of SDF-1α (red-SDF1α) was developed (Cisbio Bioassays, ref. L0012RED), whereas a red fluorescent ligand of CCK(26-33) (Red CCK(26-33), Cisbio Bioassays, ref. L0013RED) was used for the CCK1 and CCK2 binding assays.
The fluorescent ligands described in Table 1 can be found at http://www.htrf.com/products/gpcr/binding/ligands/.
Binding Data for SNAP-GPCR Fusions Using the Tag-lite Binding Assay on Adherent Cells
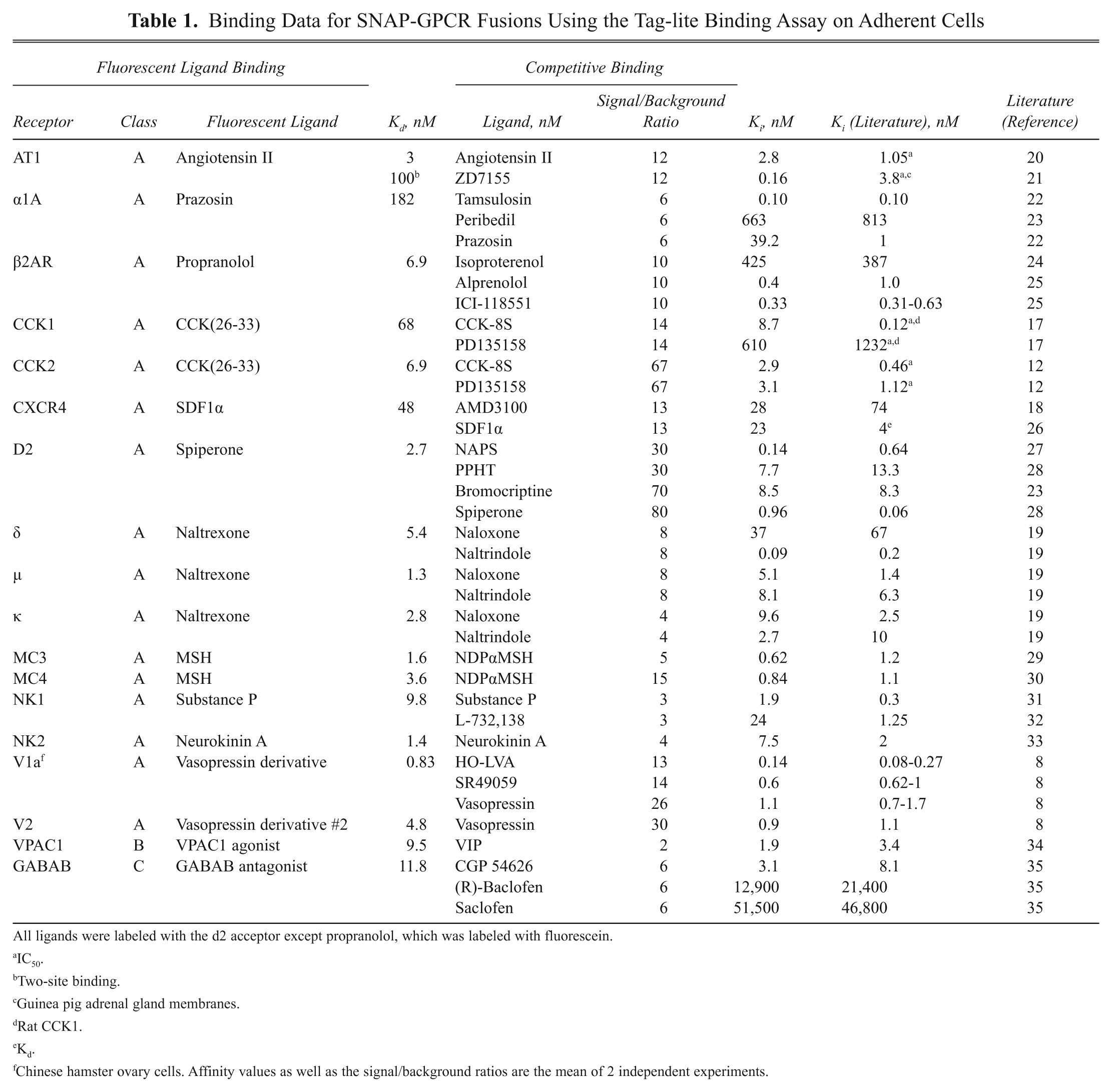
All ligands were labeled with the d2 acceptor except propranolol, which was labeled with fluorescein.
IC50.
Two-site binding.
Guinea pig adrenal gland membranes.
Rat CCK1.
Kd.
Chinese hamster ovary cells. Affinity values as well as the signal/background ratios are the mean of 2 independent experiments.
The various agonists and antagonists used for competition assays were obtained from Almac (Craigavon, UK), Tocris (Bristol, UK), Sigma (Saint Quentin Fallavier, France), or Polypeptides (Strasbourg, France). Particularly, CCK-8S, PD135158, and naltrindole were purchased from Tocris. AMD3100 and naloxone were obtained from Sigma and SDF1α from Almac.
The following SNAP-GPCR plasmids for transient transfection were used from Cisbio Bioassays: CXCR4 (PSNAPCXCR4), CCK1 (PSNAPCCK1), CCK2 (PSNAPCCK2), µ (PSNAPMOP), δ (PSNAPDOP), and κ (PSNAPOPRK1). A more extensive list, which includes all the receptors in Table 1 , can be found at http://www.htrf.com/products/gpcr/binding/plasmids/. Stable cell lines expressing the SNAP-tag-fused CXCR4 (ref. C1SU1CXCR4), CCK1 (ref. C2SU1CCK1), and CCK2 (ref. C1SU1CCK2) were developed in house as well.
Cell culture
HEK293 wild-type cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) glutaMAX (1966-021; Invitrogen, Carlsbad, CA) supplemented with penicillin 50 U/mL, streptomycin 50 µg/mL, HEPES 2 mM, 1% nonessential amino acids, and 10% fetal calf serum.
CHO-K1 cells were cultivated in F-12 HAM + glutaMAX (31765-027; Invitrogen) with penicillin 50 U/mL, streptomycin 50 µg/mL, HEPES 2 mM, and 10% fetal calf serum.
Stable cell lines were maintained in the corresponding culture medium depending on the cell background, supplemented with 0.6 mg/mL geneticin for HEK293 cell lines and 1 mg/mL geneticin for CHO-K1.
Transfection procedures
Reverse transient transfections performed on adherent cells
These transfections were performed in 96-well plates using cell density from 20,000 to 100,000 cells per well. Prior to cell plating, wells were precoated with 50 µL poly-L-ornithine for 30 min at 37°C. Transfection mixes were prepared using 200 ng of the SNAP-tag-GPCR plasmid (Cisbio Bioassays), 0.8 µL of lipofectamine 2000 (Invitrogen), and 50 µl optiMEM culture medium per well. Prior to their addition in plates, transfection mixes were preincubated for 20 min at room temperature. Then, 100 µL of HEK293 cells at a density of 1 million/mL was added in each well. Plates were incubated at 37°C under 5% CO2 for 24 h.
Transient transfections performed in batches to further generate frozen cells
Ten million HEK293 cells in complete cell medium were grown in a T1 75-cm2 flask placed at 37°C under 5% CO2. When reaching 80% confluency, cell medium was removed and replaced by 12 mL of fresh cell culture medium. In parallel, a transfection mix containing 20 µg plasmid, 60 µl lipofectamine 2000, and 8 mL optiMEM final volume was incubated for 20 min at room temperature (RT) prior to being added on cells. The culture flask was incubated at 37°C under 5% CO2 for 24 h.
Covalent labeling of cells expressing SNAP-tag®-GPCR
Adherent cells (transient and stable cell lines)
Cell culture medium was removed from the 96-well plates, and 100 nM of SNAP-Lumi4-Tb, previously diluted in the Tag-lite labeling medium, was added (100 µL per well) and further incubated 1 h at 37 °C under 5% CO2. The excess of SNAP-Lumi4-Tb was removed by washing each well 4 times with 100 µL of Tag-lite labeling medium.
Batch labeling and frozen cells (transient and stable cell lines)
After removal of the cell culture medium from a flask containing 10 million adherent cells, 10 mL of Tag-lite labeling medium containing 100 nM of SNAP-Lumi4-Tb was added to the flask and incubated for 1 h at 37 °C under 5% CO2. Cells were then detached using 5 mL of GIBCO enzyme-free Hank’s-based cell dissociation buffer (ref. 13150-016; GIBCO, Carlsbad, CA) and collected in a vial. The excess of SNAP-Lumi4-Tb was removed by 3 centrifugation/washing steps (5 min at 1500 rpm) using 10 mL of Tag-lite labeling medium. Pelleted cells were suspended in a cell culture medium containing 10% DMSO, distributed at 3 million cells per vial, and slowly frozen to −80 °C in an isopropanol-containing box. Prior to their use, the frozen cells were thawed quickly at 37 °C, the medium was removed from the vial, and the cells were suspended in the Tag-lite® labeling medium at a cell density of 0.2 to 1 million/mL depending on the cell density used in the binding assays.
Fluorescent ligand-binding assays
Using adherent cells in 96-well plates, the cell density was from 20,000 to 100,000 cells per well, whereas densities from 2,000 to 10,000 cells per well were used to carry out binding assays in suspension in 384-well plates.
Affinities of the fluorescent ligands for the receptors were determined by incubating the cells at RT with increasing concentrations of fluorescent ligand. For each fluorescent ligand concentration, the nonspecific binding was determined by adding an excess of the appropriate unlabeled compound (1 µM naltrexone for µ, δ, and κ; 10 µM CCK-8S for CCK1 and CCK2; and 1 µM AMD3100 for CXCR4). Both fluorescent ligands and unlabeled compounds were diluted in the Tag-lite labeling medium. In plates containing labeled cells and 50 µL (or 10 µL in 384-well plates) of Tag-lite labeling medium, 25 µL (5 µL for 384-well plates) of unlabeled compound or Tag-lite labeling medium was added, followed by the addition of 25 µL (or 5 µL in 384-well plates) of fluorescent ligand. Plates were then incubated at RT for 1 to 4 h before signal detection.
Competition binding assays
Both fluorescent ligands and compounds to be tested were diluted in Tag-lite labeling medium. A fixed concentration of fluorescent ligand was used (6.25 or 12.5 nM red-SDF1α for CXCR4; 10 and 30 nM red-CCK(26-33) for CCK2 and CCK1, respectively; 8 nM red-naltrexone for µ and δ; and 5 nM red-naltrexone for κ-opioid) in presence of increasing concentration of compounds to be tested. In the plates containing labeled cells and 50 µL (or 10 µL in 384-well plates) of Tag-lite labeling medium, 25 µL (or 5 µL in 384-well plates) of compound to be tested was added prior to the addition of 25 µL (or 5 µL in 384-well plates) of fluorescent ligand. Plates were then incubated at RT for 1 to 4 h before signal detection.
Signal detection and data analysis
Signal detection was performed on various fluorescent readers, Rubystar (BMG Labtech, Offenburg, Germany) or Infinite F500 (Tecan, Männedorf, Switzerland), using standard HTRF settings. When using red acceptors, the signal was collected both at 665 nm and 620 nm, whereas both 520 nm and 620 nm were used when using a green acceptor. HTRF ratios were obtained by dividing the acceptor signal (665 or 520 nm) by the donor signal (620 nm) and multiplying this value by 10,000.
Data obtained were then analyzed using GraphPad Prism (GraphPad Software, Inc., San Diego, CA) or Igor Pro (WaveMetrics, Lake Oswego, OR). Kd values of the fluorescent ligands were obtained from saturation curves of the specific binding. Specific binding was determined by subtracting the nonspecific HTRF ratio from the total HTRF ratio. Ki values of the compounds were determined from competitive binding experiments according to the Cheng and Prusoff equation. 17 Signal-to-background (S/B ratio) calculations were performed by dividing the mean of the maximum value (µmax) by that of the minimum value (µmin) obtained from the sigmoid fits.
Results
Tag-lite® binding assays on adherent cells
To validate our assay concept, fluorescent derivatives (agonists or antagonists) targeting 18 different GPCRs were designed and synthesized ( Table 1 ). The corresponding SNAP-tag fusion GPCRs have been prepared and transiently or stably expressed in different cellular backgrounds ( Table 1 ). After labeling of the adherent cells with SNAP-Lumi4-Tb, binding assays were carried out in 96-well plates. Robust HTRF signals were obtained for all GPCRs studied ( Table 1 ). Reference compounds tested in displacement assays showed consistent inhibition constants in the same range as the one measured using radioligand assays. Six of these GPCR binding assays are discussed in more detail hereafter.
CCK1 and CCK2 binding assays using adherent cells
A nonselective fluorescent agonist was synthesized to set up the assays. Briefly, the CCK(26-33) peptide was derivatized with our red d2 dye (red-CCK(26-33)). This fluorescent ligand was expected to display a good affinity for both SNAP-tag-fused CCK1 and CCK2 receptors. Its binding properties were first established using HEK293 cells transiently transfected with either SNAP-tag-CCK1 or SNAP-tag-CCK2 receptors and labeled with SNAP-Lumi4-Tb. Upon the addition of increasing concentrations of the red-CCK peptide, the HTRF signal between Lumi4-Tb and the red fluorescent acceptor, expressed in the HTRF ratio, increased in a dose-dependant manner (
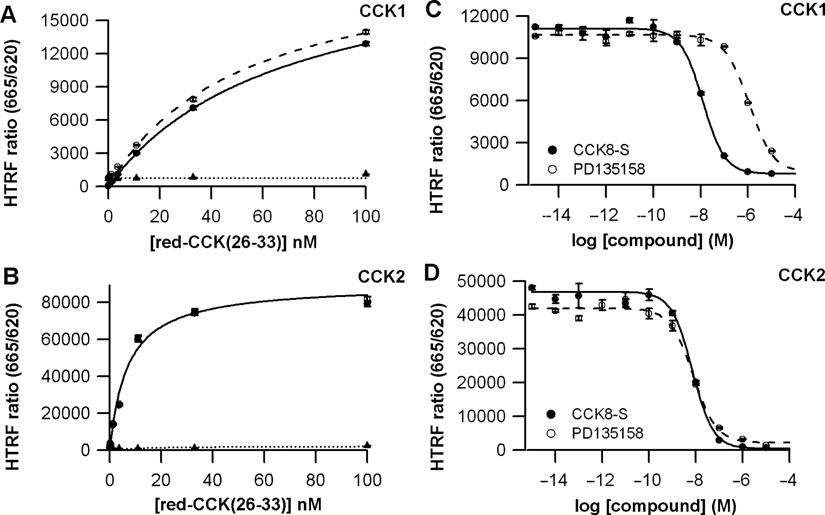
CCK1 and CCK2 ligand-binding assays. (
Competitive binding assays were set up by using 2 competitors, the sulfated CCK-8 (CCK-8S) as an agonist and a nonpeptidic antagonist, PD135158. They were carried out using either 30 nM of the fluorescent ligand in the CCK1 assay or 10 nM in the CCK2 assay.
Since it can be more convenient for HTS campaigns, both SNAP-tag-CCK1 and SNAP-tag-CCK2 stable cell lines were generated in a CHO-K1 and HEK293 cellular background. As shown in Table 2 , the binding data obtained for these stable cell lines are consistent with those previously obtained with transiently transfected cells. Particularly, the selectivity of PD135158 for CCK2 is maintained, as well as the high potency of the CCK-8S agonist for both receptors.
Comparison of Binding Data Obtained with Either Transiently Transfected Cells or Stable Cell Line Expressing SNAP-tag-CCK1 or SNAP-tag-CCK2
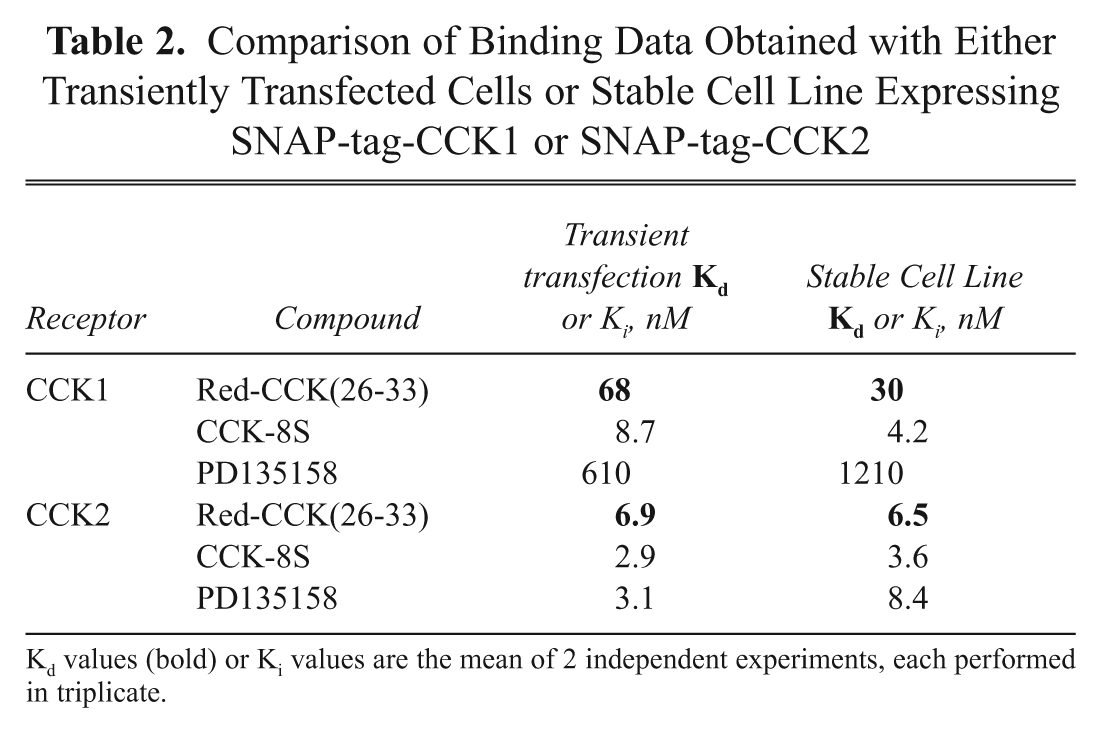
Kd values (bold) or Ki values are the mean of 2 independent experiments, each performed in triplicate.
CXCR4 binding assay using adherent cells
A fluorescent derivative of SDF-1α/CXCL12 (red-SDF1α) was synthesized using the d2 dye as previously described for a Texas red derivative. 16 A HEK293 stable cell line expressing the SNAP-tag-fused CXCR4 receptor (ST-CXCR4) was established to carry out the binding assays in 96-well microplates. Optimal binding parameters using red-SDF1α were obtained with a cell density of 20,000 cells/well. The HTRF signal, expressed as the HTRF ratio, turned out to follow a standard 1-site binding curve upon addition of increasing concentrations of red-SDF1α ( Fig. 3A ). The nonspecific binding was determined using an excess of the nonpeptidic antagonist AMD3100 (5 µM). A Kd value of 48.0 ± 5.1 nM was obtained for the red-SDF1α, similar to that previously obtained for the Texas red derivative. 6 Competitive binding assays were carried out using a nonpeptidic antagonist AMD3100 and unlabeled SDF1α as competitors. As expected, Figure 3B shows a decrease of the HTRF signal upon addition of increasing concentration of the 2 compounds, clearly demonstrating the displacement potencies for these 2 competitors. Typical dose-response curves were obtained, leading to pIC50 values of 7.45 ± 0.12 and 7.53 ± 0.06 for AMD3100 and SDF-1α, respectively. The resulting Ki values of 28 and 23 nM are in good agreement with values published in the literature. 18 It establishes this homogeneous binding assay as a new screening solution to identify compounds targeting the CXCR4 receptor.
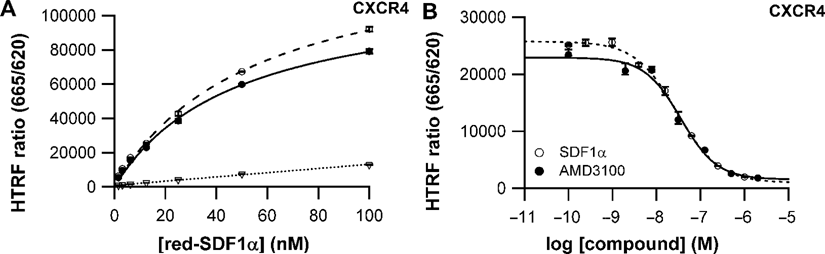
Ligand-binding assay for CXCR4 using adherent cells. (
Opioid receptor binding assays using adherent cells
To develop Tag-lite binding assays covering the 3 subtypes of opioid receptors, µ, δ, and κ, the potent nonselective antagonist naltrexone was chosen as the parent ligand to design a fluorescent derivative. This nonpeptidic compound was derivatized with the d2 dye to generate naltrexone-red. The affinity of naltrexone-red for the 3 receptor subtypes was established using HEK293 cells transiently transfected with SNAP-tag-µ-opioid, SNAP-tag-δ-opioid, or SNAP-tag-κ-opioid receptors as described previously for other GPCRs. The nonspecific assay signal was determined by using 10 µM of unlabeled naltrexone. As expected, this fluorescent ligand displayed a good affinity for each opioid receptor subtype, leading to robust HTRF signals. Kd values obtained were 1.3 ± 0.4 nM, 5.4 ± 1.8 nM, and 2.8 ± 0.6 nM for µ, δ, and κ receptors, respectively. For competition assays ( Fig. 4 ), naltrexone-red was incubated in the presence of increasing concentrations of 2 antagonists, naloxone and naltrindole. Ki values for naloxone were determined to be 5.1 nM, 37 nM, and 9.6 nM for µ, δ, and κ receptors, respectively ( Fig. 4B ). These data indicate that this compound is a potent nonselective opioid antagonist and are in good agreement with literature values. 24 Ki values for naltrindole were 8.1 nM, 0.09 nM, and 2.7 nM for µ, δ, and κ, respectively, indicating a good δ/µ or κ selectivity ratio for this compound ( Fig. 4A ). This is also in good agreement with data previously published. 19 The above results strongly suggest that the established Tag-lite opioid receptor binding assays can be a useful tool to profile any compound selectivity against the different opioid receptor subtypes. Since they are carried out using a single fluorescent ligand, they can easily be run in parallel for HTS or profiling purposes.
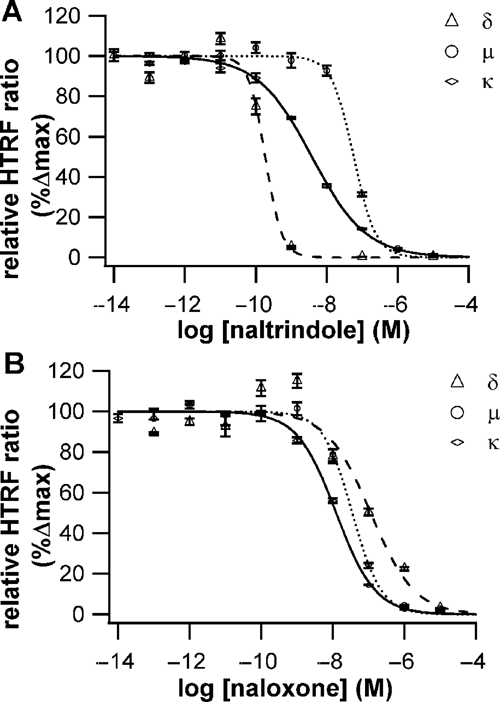
Pharmacological profiling of naltrindole and naloxone on the δ, µ, and κ opioid receptor using naltrexone-red as a tracer. (
Ligand-binding assays using cells in suspension
After the validation of a ligand-binding assay using adherent cells, an alternative assay format using prelabeled cells in suspension was developed to simplify miniaturization and automation. Cells expressing either transiently or stably a SNAP-tag-GPCR fusion protein were labeled in batch quantities with SNAP-Lumi4-Tb and frozen at −80°C for long-term storage. To carry out a Tag-lite binding assay, samples of frozen cells were thawed just before use and dispensed together with their corresponding fluorescent ligand and a set of compounds to be tested ( Fig. 1C ). This mix-and-measure assay format is therefore based on cells in suspension. It was first validated using both SNAP-tag-CXCR4 and SNAP-tag-CCK2 stable cell lines.
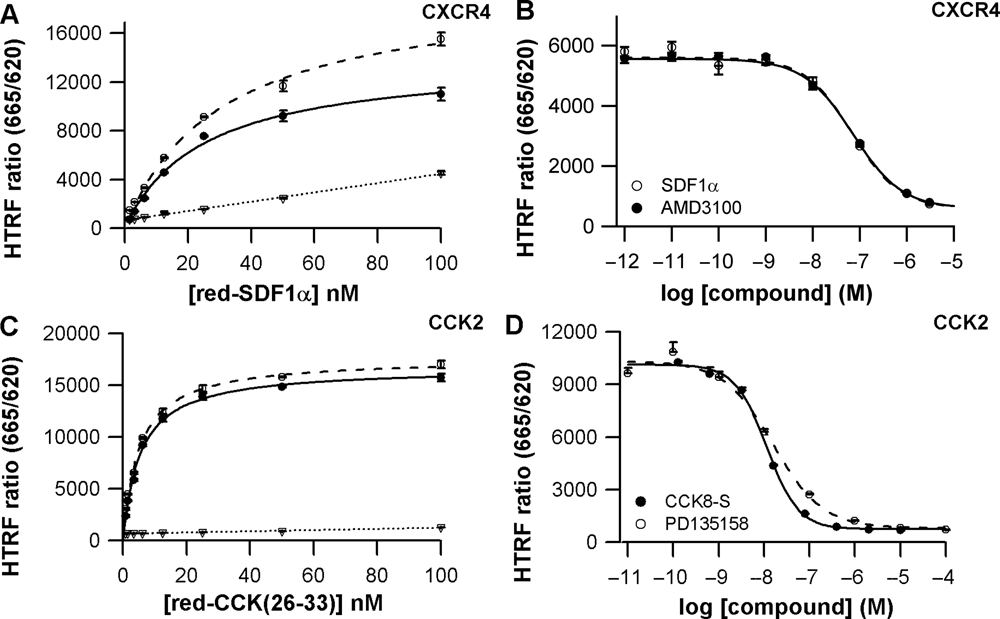
Homogeneous ligand-binding assays on stable cell lines expressing CXCR4 or CCK2 cells used in suspension. (
Comparing Binding Data for SNAP-tag GPCR Fusion Using the Tag-lite Binding Assay with Adherent Cells or Cells in Suspension
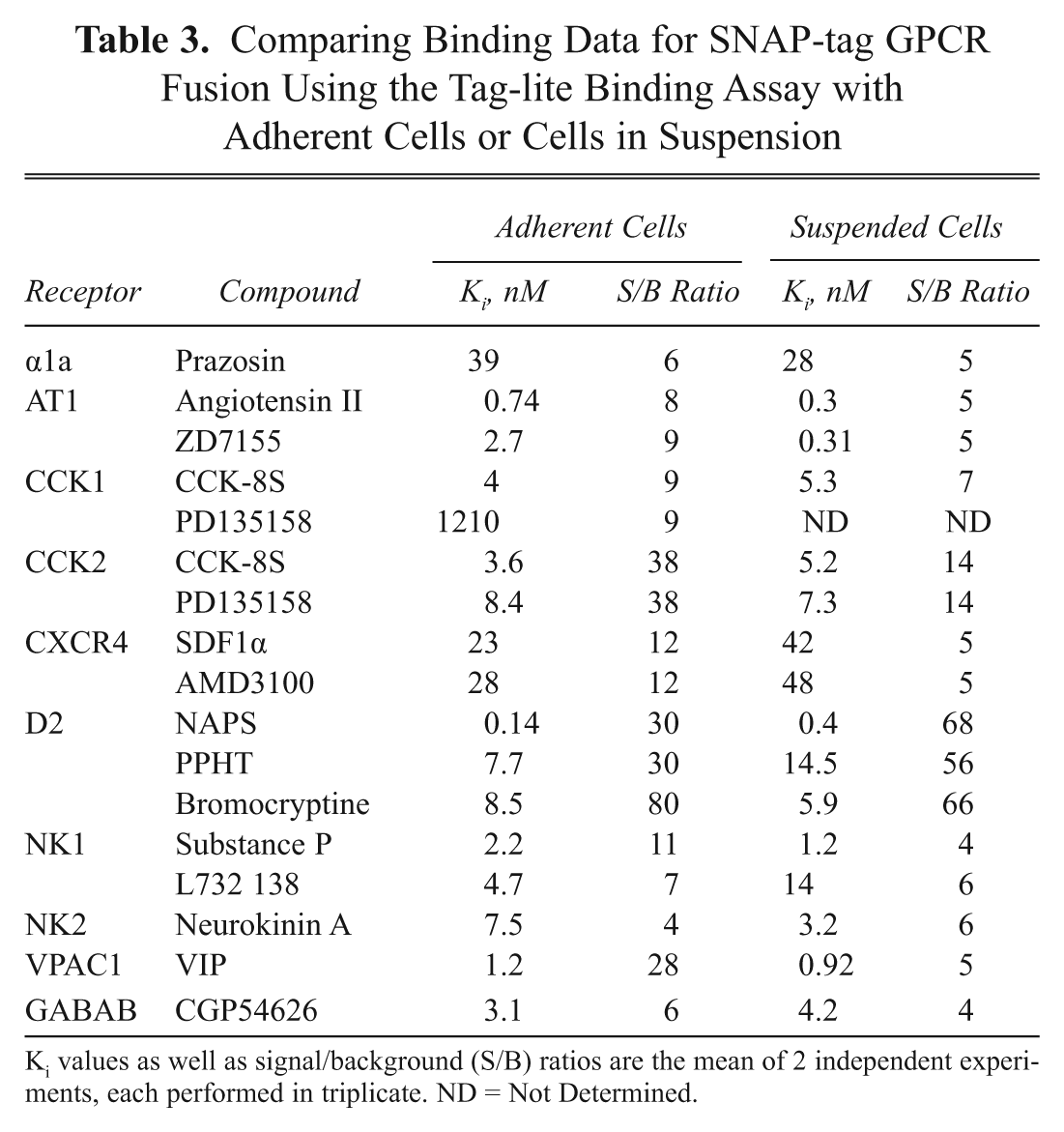
Ki values as well as signal/background (S/B) ratios are the mean of 2 independent experiments, each performed in triplicate. ND = Not Determined.
Since preliminary results with this new assay format were very encouraging using stable cell lines as starting material, it was extended to test transiently transfected cells. For that purpose, a series of 4 plasmids coding for SNAP-tag-fused α1a-adrenoceptor, dopamine D2, neurokinin NK2, or GABA-B receptors was transfected into HEK293. After 24 h of transfection, cells were labeled with SNAP-Lumi4-Tb and frozen at −80°C. After thawing, viable labeled cells were used to carry out the different Tag-lite binding assays. Table 3 compares the results obtained using cells in suspension to adherent cells. In general, the output of the GPCR binding assays is consistent whatever the assay format considered, validating the use of frozen prelabeled cells obtained from transient transfections as a ready-to-use cellular reagent to carry out mix-and-measure Tag-lite binding assays.
Discussion
This study, carried out on a large panel of GPCRs, establishes the Tag-lite technology as an attractive alternative to other GPCR binding techniques. Based on SNAP-tag-fused receptors, binding assays for GPCRs from different classes were successfully established. In particular, a nonradioactive GABA-B binding assay has been established for the first time. As previously shown for class C GPCRs, 11 this study suggests that the binding properties of GPCRs from classes A and B are not dramatically modified by their N-terminal tagging with the SNAP-tag enzyme.
Tag-lite binding assays require the availability of fluorescent ligands. In most cases, we succeeded in developing peptidic and nonpeptidic fluorescent ligands based on a red acceptor that displayed a good affinity for the different targets. A single exception was met with the β2-adrenergic receptor, for which only a green acceptor derivative was found to be active. It suggests that the chemical structure of the red acceptor impairs the binding properties of propranolol. On the opposite, red derivatives of spiperone and naltrexone displayed high affinities for dopamine D2 and opioid receptors, respectively. It underlines how, when starting from a low molecular weight compound, the design of a fluorescent ligand can be challenging. The chemical features of the fluorescent probe, the position of derivatization used to label the parent compound, and the nature of the linker between the 2 moieties are key parameters to take into account. In that context, the possibility to use either a green or a red dye to design a Tag-lite ligand is a great advantage to maximize the chances of success. However, one can speculate that designing some particular types of ligand-like lipids, which offer limited possibilities of derivatization, will be particularly challenging.
Like the other FRET-based techniques, Tag-lite minimizes nonspecific binding detection and therefore ensures the assay signal specificity. This is of particular interest when investigating highly hydrophobic compounds or ligands, such as SDF1α, that possess other binding sites at the plasma membrane. 16 Moreover, the use of a TR-FRET detection gives robust S/B ratios even in a miniaturized format.
Tag-lite allows the determination of both Kd values of the fluorescent ligands and Ki values of the investigated compounds. Values obtained in this study using reference agonists or antagonists are consistent with those obtained using standard radioactive techniques ( Table 1 ). To definitely assess the relevance of the Tag-lite binding assays, further validations based on a larger panel of compounds may be required on some specific targets.
However, like the other FRET-based ligand-binding technologies, Tag-lite does not allow the determination of the cellular receptor density. The specific signal obtained in these techniques is the product of the concentration of bound ligand and the efficiency of the FRET process. Moreover, the signal cannot be linked to any free signal since unbound ligands do not generate any FRET. This makes the calculation of the cellular receptor density extremely difficult and practically impossible to perform.
When carried out on adherent cells, Tag-lite binding assays require several washing steps performed into plates and are therefore not well adapted to miniaturization and automation. SNAP-Lumi4-Tb labeling of membrane preparations prior to their freezing for a further use was considered an option to make the assay more HTS-friendly. However, despite positive preliminary results obtained on the V1a receptor (data not shown), we decided to focus our efforts to develop batches of prelabeled frozen cells. Since it does not require any membrane preparation, such a process was easier to establish and ensures a better batch-to-batch reproducibility. After thawing, prelabeled cells can be used straightforward in suspension in a homogeneous format of the binding assay. It can be performed with only a few thousand cells per well in 384 wells, and a further miniaturization in a 1536-well format can be easily envisaged. It is therefore an attractive alternative to the other fluorescence-based techniques to carry out binding assays in primary screenings, making this assay suitable for ultra-HTS (uHTS) campaigns. S/B ratios are better than those usually obtained with FP or FRET-based techniques and compare well with those obtained using DELFIA, a technique more complicated to miniaturize and automate.
Since oligomerization assays 11 or functional assays are also available through the Tag-lite technology, their combination with this newly established binding assay format might be considered, opening the route to a multidimensional characterization of GPCR structures and functions. Using the same cell line expressing a SNAP-tag GPCR fusion protein, it is therefore possible to run several assays to deeply characterize compounds of interest and better understand their mechanisms of actions. Key information about their binding mode, as well as their effects on various signaling pathways, membrane organization features, and the GPCR recycling process, can be obtained in an HTS-compatible format. Further Tag-lite developments should also offer the possibility to combine in a single well a binding assay with downstream effector measurements. Since 2 different acceptors are available to create FRET pairs with Lumi4-Tb, such a multiplexed assay may be achievable through the Tag-lite technology.
Footnotes
Acknowledgements
We thank Françoise Baleux, Angélique Levoye, and Françoise Bachelerie from the Pasteur Institute in Paris for valuable discussions regarding the function and binding of SDF-1α on the CXCR4 receptor.
HTRF® and Tag-lite® are registered trademarks of Cisbio Bioassays. Lumi4® is a registered trademark of Lumiphore, Inc. DELFIA® is a registered trademark of PerkinElmer, Inc. SNAP-tag® is a trademark of News England Biolabs, Inc. Taglite ligands for the β2AR and GABA-B receptors were created exclusively for Cisbio by CellAura Technologies