
Abstract
Fragment-based drug discovery (FBDD) has become a widely accepted tool that is complementary to high-throughput screening (HTS) in developing small-molecule inhibitors of pharmaceutical targets. Because a fragment campaign can only be as successful as the hit matter found, it is critical that the first stage of the process be optimized. Here the authors compare the 3 most commonly used methods for hit discovery in FBDD: high concentration screening (HCS), solution ligand-observed nuclear magnetic resonance (NMR), and surface plasmon resonance (SPR). They selected the commonly used saturation transfer difference (STD) NMR spectroscopy and the proprietary target immobilized NMR screening (TINS) as representative of the array of possible NMR methods. Using a target typical of FBDD campaigns, the authors find that HCS and TINS are the most sensitive to weak interactions. They also find a good correlation between TINS and STD for tighter binding ligands, but the ability of STD to detect ligands with affinity weaker than 1 mM KD is limited. Similarly, they find that SPR detection is most suited to ligands that bind with KD better than 1 mM. However, the good correlation between SPR and potency in a bioassay makes this a good method for hit validation and characterization studies.
Keywords
Introduction
P
The use of fragments as starting points for drug discovery has a number of advantages. Fragments make better use of both the scaffold and functional groups to bind the target with the result that their ligand efficiency (LE, defined as ΔGbind/heavy atom count 4 ) is significantly higher than that of compounds found by HTS. 5 Higher LE allows increased flexibility when elaborating hits and critically allows for a smaller lead compound. 6 Furthermore, small-molecule fragments possess better complementarity to target proteins, resulting in a higher hit rate than HTS, 1 and because the available chemical space is many orders of magnitude smaller, it can be much more efficiently sampled. As a result, FBDD requires a small but rationally designed library that can be carefully controlled to ensure quality. However, fragments, due to the limited potential to productively interact with the protein, typically bind with KD > 10 µM. Such low-affinity hits can only be identified using highly sensitive biophysical methods such as high-throughput X-ray crystallography, 7 nuclear magnetic resonance (NMR), 8 and surface plasmon resonance (SPR) 9 or, when appropriate, an enzyme inhibition assay performed at high concentration of the fragment. 10 NMR screening methods range from the detection of signals from 15N- or 13C-labeled target proteins 8 to so-called ligand-observed methods, which monitor the 1H or 19F spins of the fragments. 11,12 However, because NMR remains a mass insensitive technique, screening of even relatively small fragment libraries still requires 10s to 100s of mg of purified protein. Recently, we developed a screening method called target immobilized NMR screening (TINS), in which a target and a reference protein are immobilized on solid media, and binding of fragments to the immobilized proteins is monitored by NMR. 13 One advantage of this method over others is the small amount of protein required because TINS uses a single sample of the target to screen an entire fragment library. 14
Because fragments are by definition small, even those that bind the target very weakly may be characterized by high LE. For example, a fragment containing 12 heavy atoms with a KD of 2 mM has an LE > 0.31, a generally accepted threshold for a desirable starting point for elaboration. 4 Therefore, the ligand screening technique must be capable of detecting weak binding well into the single-digit mM range. On the other hand, the technique should be robust to avoid a high false-positive rate. Here we present a comparison study of TINS to the fragment screening techniques most commonly used in FBDD—namely, saturation transfer (STD) difference NMR spectroscopy, 11 SPR, and high-concentration bioassay. The viral RNA-dependent RNA Polymerase (RdRP) was selected as a target because it is both typical of an FBDD project and amenable to all assay formats. 15 A fragment library was initially screened against RdRP using TINS. Based on this screen, 133 fragments were selected, including both hits and nonhits, and rescreened using STD. The hits generated by both methods were characterized in a bioassay for inhibition of RdRP activity. A subset of fragments that were positive in all 3 assays was then characterized for binding to RdRP using an SPR biosensor assay. On the basis of these results, we provide recommendations for the optimal use of each method.
Materials and Methods
Fragment preparation for TINS and 1D-STD
The composition of the fragment library has been described. 16 The molecular weight of the fragment library ranged from 150 to 300 Da and averaged 220 Da. Compounds were dissolved in DMSO-d6 at 50 or 100 mM and were subsequently diluted to 500 µM in the final aqueous buffer solution used for screening. The fragment mixes consisted of typically 3 to 5 fragments. For both STD and TINS screening, fragment mixes were prepared in screening buffer, which comprised 25 mM Tris-d11 (pH 7.5), 150 mM NaCl, and 1 mM MgCl2 in 99.9% D2O.
Target preparations
The viral RdRP, which included a C-terminal 6 His-tag (63 kDa), was expressed in Escherichia coli strain BL21 DE3+ and purified using a combination of nickel affinity and cation exchange chromatography. The C-terminally 6 His-tagged PH domain (aa 1-123) from human Akt1 (15 kDa) was expressed using BL21 DE3+ cells and purified using a similar procedure for RdRP.
High-concentration bioassay
Polymerase activity was assayed as described. 15 The percentage of inhibition (PIN) per reaction was calculated as follows:
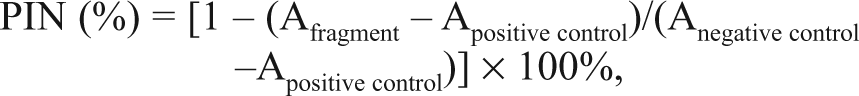
where Afragment and Anegative control are the radioactivity counts measured in the presence of the fragment and DMSO, respectively, whereas Apositive control is the radioactivity measured in the presence of a known inhibitor that abrogates enzymatic activity completely. The assay was performed in duplicate for samples and in triplicate for the negative (0.5% or 2% DMSO) control and the positive control in the presence of a known inhibitor at 250 µM. The quality of the assay was monitored by Z′ score calculated from the controls. 17
TINS methodology and analysis
In total, 4 mg of the viral protein and 1 mg of Akt1 PH domain were immobilized via amine coupling to 500 µL (bed volume) of Actigel-ALD resin (Sterogene, Carlsbad, CA) according to the manufacturer’s protocol in 50 mM Na phosphate (pH 7.4) and 150 mM NaCl solution overnight at 4°C. Typically, an immobilization efficiency of 90% was achieved using this protocol, and the final concentration of immobilized protein was estimated to be in the range of ~110 µM. Unreacted sites on the resin were blocked by addition of 100 mM d11-Tris. The resin was then packed into a dual-cell sample holder. 14
TINS NMR measurements were performed as described 14 with the exception that a spatially selective Hadamard pulse sequence 18 was used. A T2 delay of 80 ms using a CPMG sequence was added prior to signal acquisition to suppress resonances arising from the resin. The change in signal amplitude due to interaction of the fragment with either target or reference protein is referred to as the TINS effect and was calculated as a weighted average of the TINS effect observed for each unique signal (n) of a fragment:
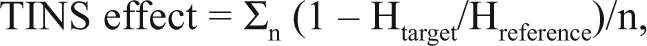
where H is the height of the observable signals. The amount of solution in the target and reference cells can be determined by injecting the TINS running buffer in H2O into each cell and integrating the signal. In general, the difference in the integrated volume of water signal in the 2 cells must be below 10%. This difference is reflected in the difference of the height of resonances of nonbinding reference molecules (0.5 mM imidazole, 0.5 mM Na acetate, 4% d6-DMSO, and 0.5 mM TMA) between the 2 cells. Therefore, 0.1 was used as the lowest cutoff value for a significant TINS effect in the hit identification process.
STD methodology and analysis
All STD NMR measurements were performed using a Bruker AV 500-MHz spectrometer (Bruker, Billerica, MA) equipped with a TXI probe. The purified viral protein was used at 20 µM, and the fragment was assessed using the same mix and conditions as used in the TINS experiment. The STD spectrum was recorded using the pulse scheme reported previously. 11 The total number of scans was 512, resulting in a 54-min experiment time per sample. The STD effect was calculated as follows:
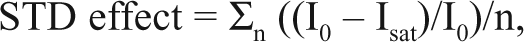
which expresses the signal intensity (Isat) in the on-resonance spectrum as a fraction of the intensity of the unsaturated off- resonance spectrum averaged over all observable signals. A cutoff of 0.02 (2 times the standard deviation) for a significant STD effect was used for hit identification in the screen.
SPR biosensor methodology
The interaction between the viral enzyme and validated fragment hits was studied on a Biacore T100 instrument (GE Healthcare, Piscataway, NJ), equilibrated at 25°C. CM5 sensor chips (research grade; GE Healthcare) were used for the experiments. Viral enzyme sensor surfaces and reference surfaces were prepared by standard amine coupling via exposed primary amines (amine coupling kit; GE Healthcare). Typical immobilization levels ranged from 9500 to 12,500 resonance units (RU) and 1800 to 2200 RU, respectively. Surfaces were stable for at least 4 days, as measured by Rmax and the signal-to-noise ratio.
For interaction studies between the immobilized viral protein and the fragments, 25 mM Tris (pH 7.5), 150 mM NaCl, 1 mM MgCl2, 0.005% of the detergent P20 (GE Healthcare), and 0.1% or 0.4% DMSO were used as running buffer. Stock solutions of the fragments were made in DMSO (10 mM). Samples were injected for 60 s with a flow rate of 30 µL/min in a series of 4 concentrations with a maximum of 200 µM. To determine specific binding, the reference channel, containing the immobilized Akt1 PH domain, was subtracted from the experimental channel.
Results
Screen of the fragment library for binding to RdRP using TINS
The viral and reference proteins were immobilized via primary amines, and the functionality of immobilized RdRP was determined using the bioassay described. Subsequently, the immobilized proteins were packed into a dual-cell sample holder.
14
Due to susceptibility mismatch between the sepharose beads and surrounding aqueous buffer, the line width of small molecules increases from about 1 to 11 Hz when recorded at 500 MHz. This line broadening is specifically taken into account when designing mixtures of fragments so that every fragment has at least 3 well-resolved resonances in the spectrum. Binding of a fragment to a protein molecule immobilized on a solid support causes the resonances of a fragment to further broaden into the kHz range. As a result, ligand binding is detected by a simple reduction in the height of all NMR signals from that fragment (
Screening of the fragment library for binding to RdRP using target immobilized NMR screening (TINS). (
A library consisting of 1270 commercially available fragments was screened against the viral RdRP using TINS. The physicochemical properties of the fragment library have been reported previously. 16 The library was screened by repeated cycles of injection of mixtures of fragments, which typically consisted of 3 to 5 compounds, into both cells packed with either immobilized target or reference protein. After the mixture of compounds was injected, the flow was stopped, the NMR data were acquired, and then the fragments were washed from the cell prior to application of the next mix. Thus, a single sample of only 4 mg of viral polymerase was required for the entire fragment library screen by TINS. Based on the initial TINS screening data, a total of 30 mixtures, which consisted of 7 mixtures without binders and 23 mixtures containing at least 1 binder per mix (and many specifically selected to have more than 1), were chosen for screening by STD. The 30 mixtures contained 133 unique fragments, from which TINS had identified 74 hits using the criteria given.
1D-STD screen setup
In STD spectroscopy, one observes a difference in the amplitude of ligand resonances in 2 separate experiments, in which the magnetization of the target protein is alternately saturated (on-resonance spectrum) or unsaturated (off-resonance spectrum).
11
The attenuation of the NMR signals of a ligand occurs in the on-resonance spectrum via through-space, noncoherent transfer of saturation from the 1Hs of the target protein to those of the bound ligand. When a mixture of compounds is assayed in the presence of a target protein, the resulting difference spectrum between the on- and off-resonance saturation spectra will contain only the signals from the ligand, whereas the signals from the nonbinders are removed in the difference spectrum. The sensitivity of the STD experiment on our 500-MHz magnet equipped with a room temperature probe was optimized using a sample of human serum albumin (HSA; 66 kDa) and known binders, including acetaminophen with a KD of 690 µM, typical for fragments binding to a target.
20
Titration of HSA into 500 µM acetaminophen indicates that the size of the STD effect is linear with the protein concentration range tested (
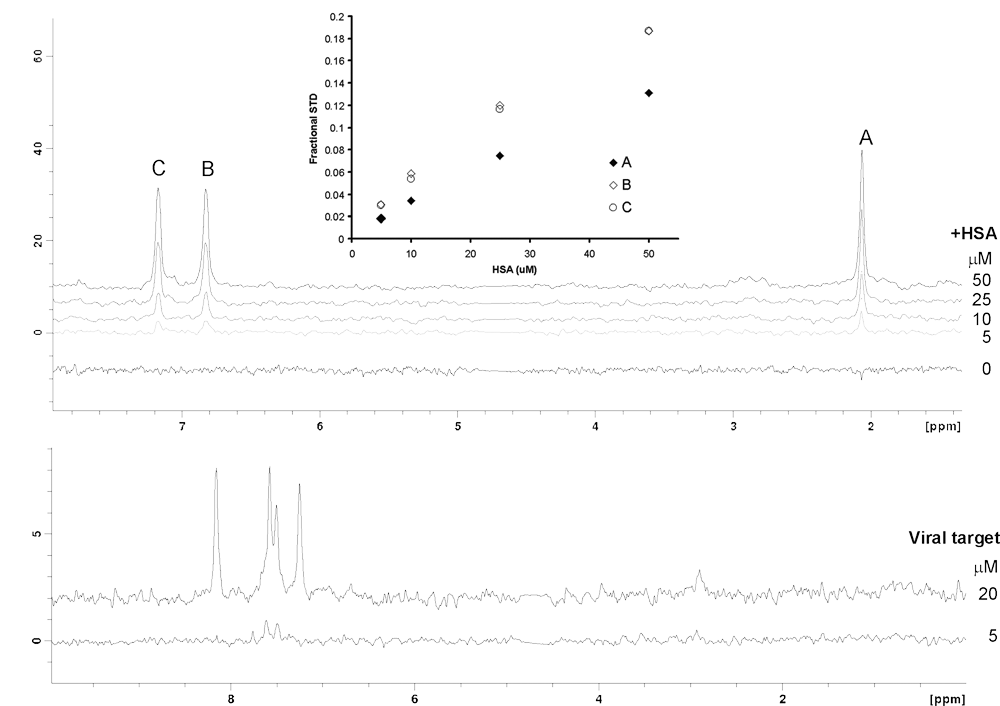
Saturation transfer difference (STD) experiment was optimized using human serum albumin (HSA) and 500 µM of acetaminophen as a control. (
Initially, the STD NMR screening was conducted at 5 µM RdRP, which resulted in far fewer hits than when the screen was performed at 20 µM. Clear improvements in the signal-to-noise (S/N) ratio of STD signals from a binding fragment were observed when the viral protein concentration was raised by 4-fold (
Comparison of TINS and STD in fragment screening
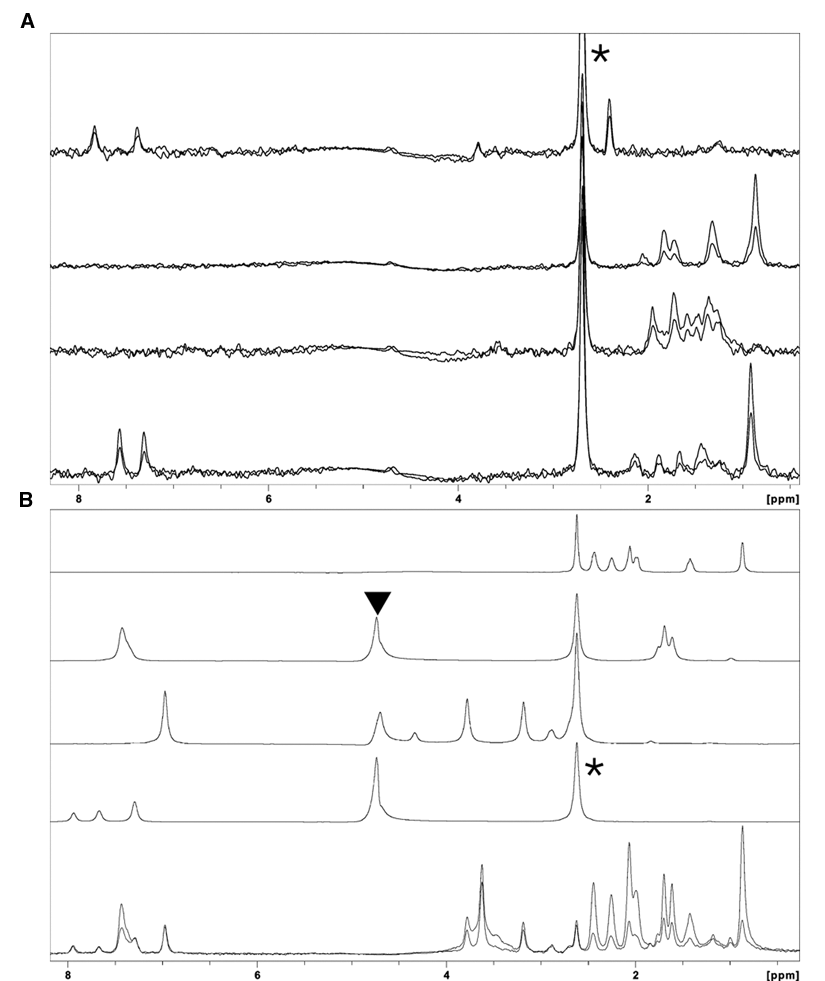
Using the criteria established, we detected 49 hits from the 133 fragments screened using STD compared to the 74 we had previously detected using TINS. Combining the unique hits from both TINS and STD gives a total of 83 hits. Examples of shared and unique hits from an identical mixture are depicted in
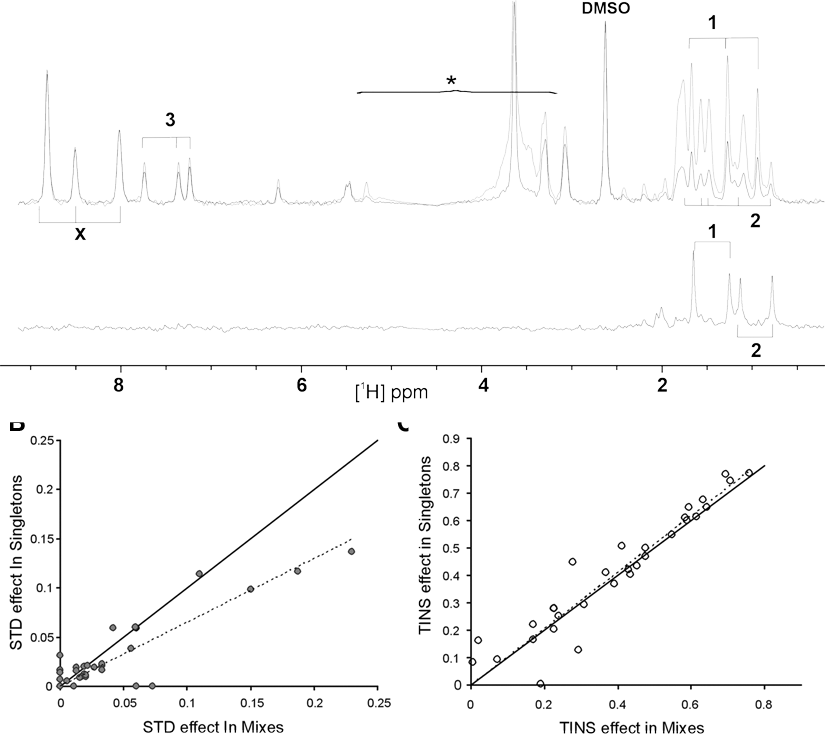
Comparison of hits identified by saturation transfer difference (STD) and target immobilized NMR screening (TINS) from a mix of fragments. (
Characterization of NMR hits by a high-concentration bioassay
High-concentration bioassays have been successfully applied to the initial stage of fragment hit identification.
21
Typically, biophysical methods (e.g., X-ray, NMR, and SPR) are used to validate bioassay hits by monitoring the physical interaction with the target. In the present study, a high-concentration bioassay was performed on the NMR hits to characterize their inhibitory activity against the RdRP polymerase. The 83 total hits were individually assayed at 500 µM in the polymerase assay. The criteria applied in classifying a fragment as an inhibitor vary significantly. In looking at only 2 examples, one finds thresholds of (using PIN) PIN >30% at the 200-µM fragment
22
and a PIN >60% at the 1.0-mM fragment.
23
Here, fragments were considered “inhibitors” when they exhibited a PIN value above 20% (group I in
Comparison of NMR to high-concentration bioassay for ligand identification
An inherent advantage of a high-concentration screening is that it can be readily quantified. We wished to assess whether either the TINS or STD effects observed correlated with the PIN at the given fragment concentration. For this purpose, we treated an activator as equivalent to an equipotent inhibitor. As shown in
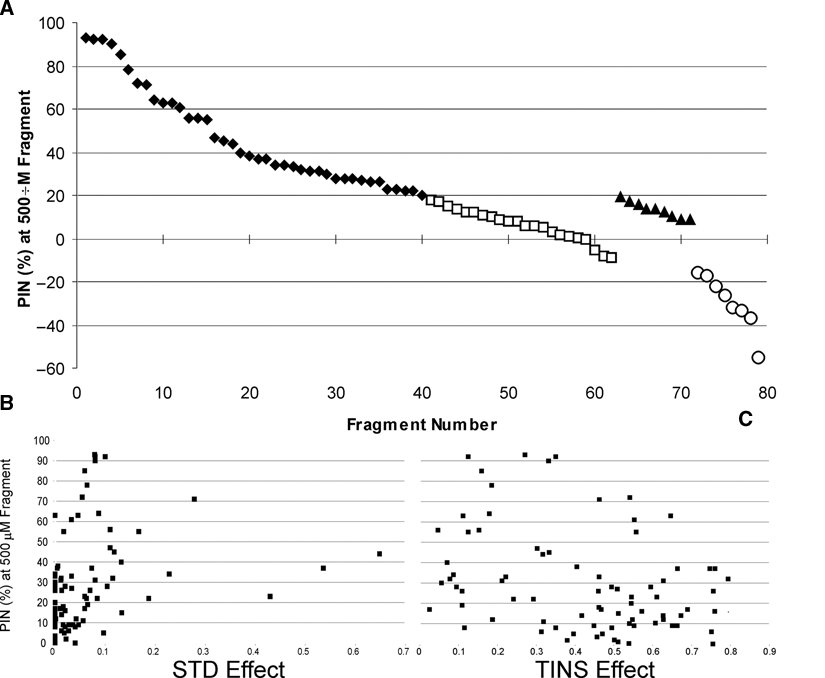
Comparison of saturation transfer difference (STD) and target immobilized NMR screening (TINS) effects to biological activity. (
A summary of results from the 3 approaches is presented in
Comparison of Hits from STD, TINS, and Bioassay

STD, saturation transfer difference; TINS, target immobilized NMR screening; PIN, percentage of inhibition; NMR, nuclear magnetic resonance.
Examples of Compounds with Results of Each Detection Method
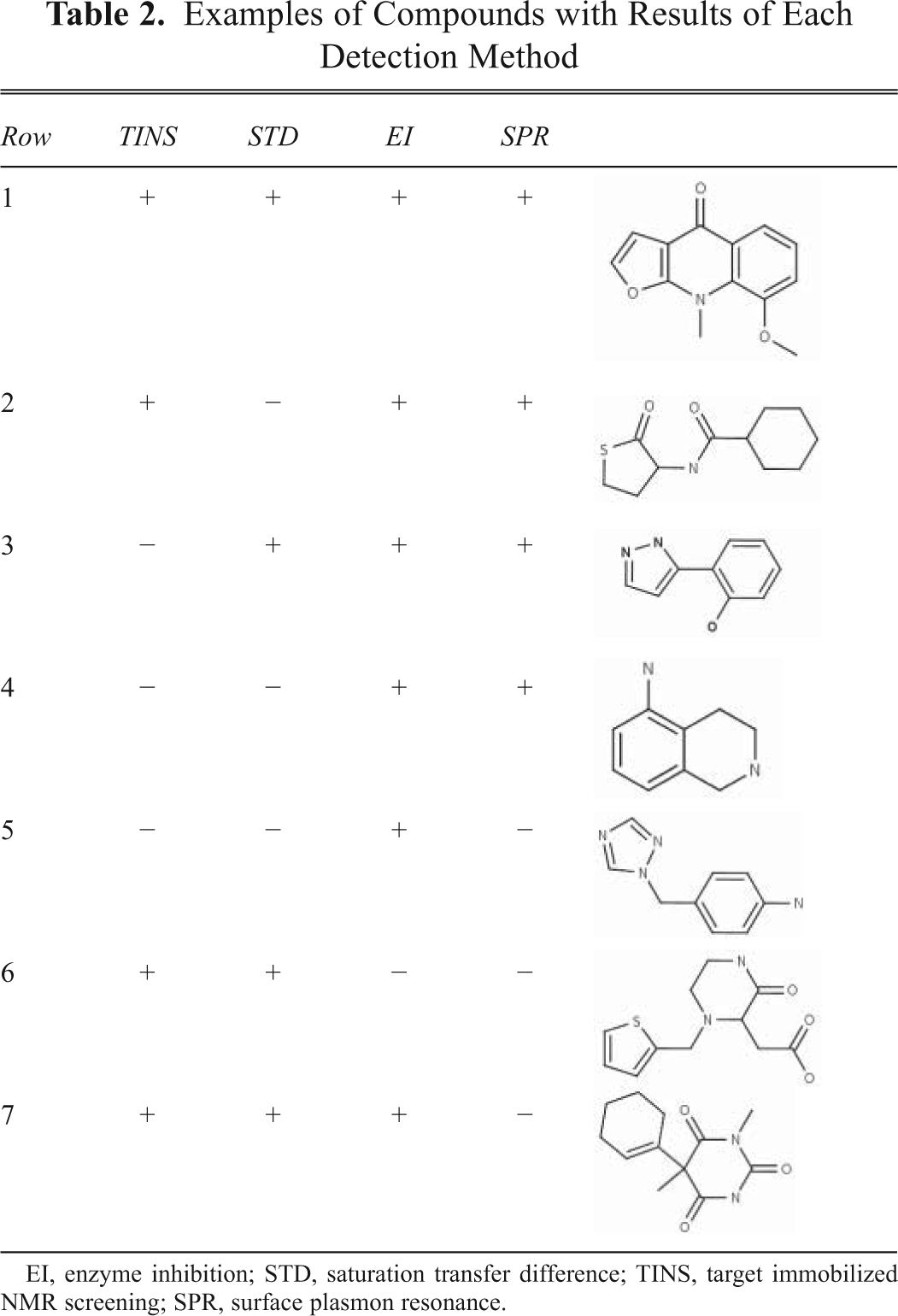
EI, enzyme inhibition; STD, saturation transfer difference; TINS, target immobilized NMR screening; SPR, surface plasmon resonance.
Fragment screen by SPR biosensor
We wished to compare the ligand screening results of the NMR and biochemical methods to those obtainable by SPR using a Biacore T100 (Biacore, Piscataway, NJ), an instrument typically employed for this purpose. As with TINS, the target is immobilized to detect binding in SPR. Subsequently, a solution of the fragment to be tested (one at a time) is flowed over the surface of immobilized target, and the change in signal detected upon binding is in general proportional to the mass of the compound and the number of available binding sites on the surface. Because fragments have a low molecular weight, high surface densities of immobilized protein are necessary. 9 A total of 62 fragments from the STD and TINS hits and 1 known ligand with a mass of 385 Da (IC50 of ~10 µM) were assayed for binding to RdRP using SPR with the Akt1 PH domain as a reference. The selected fragments consisted of 30 from group I, 23 from group II, 7 from group III, and 2 fragments that were negative in both STD and TINS but positive in the enzyme assay.
To establish the validity of our SPR assay, we first titrated the known ligand at a concentration range of 1.25 to 10 µM to establish a linear response (
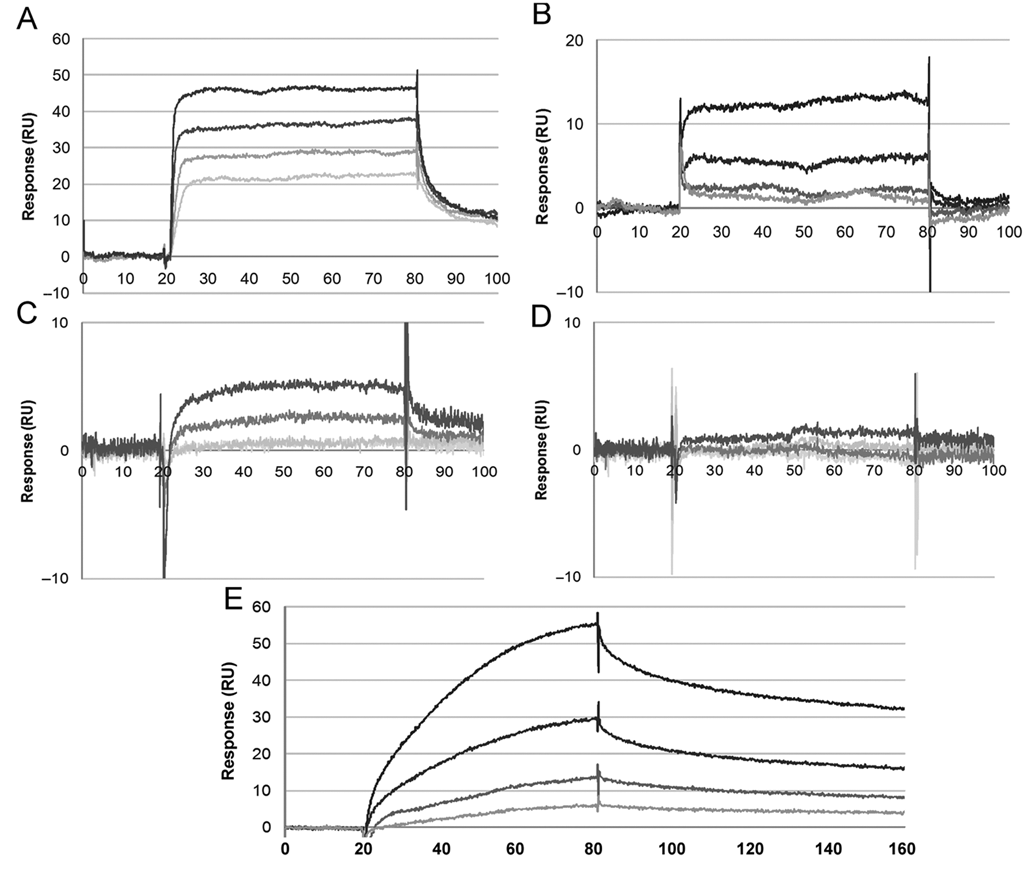
Example sensorgrams for the interaction between RdRP and test compounds. Each sensorgram depicts binding data for a concentration series of that compound. (
We compared the results of the SPR assay to the biochemical assay by grouping the SPR results in compounds that were “not detected,” “low affinity,” “high affinity,” and “positive control” according to the definitions above and plotted these against the observed inhibition in the bioassay (
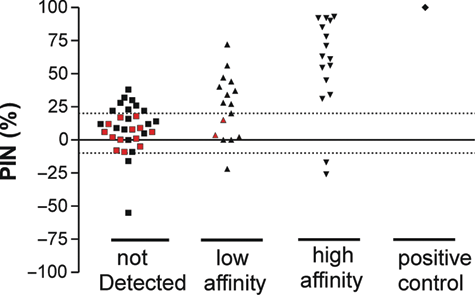
Comparison of the interaction between fragments and RdRP as measured by surface plasmon resonance (SPR) to the percentage of inhibition (PIN) obtained from the bioassay. Interactions on SPR are shown on the x-axis and divided into fragments that did not bind in the SPR assay, low and high affinity measured on SPR and the positive control. Red symbols indicate fragments from group II.
Discussion
In this present study, we compared the sensitivity and robustness of 4 different methods for primary screening of fragment libraries for binding to a pharmaceutical target: 2 different ligand-observed NMR techniques, STD and TINS; high-concentration bioassays; and SPR. There are a number of characteristics that a screening technique must encompass to be of use in FBDD. A technique should be broadly applicable to (nearly) all potential targets and sensitive to a weak binding well into the mM range while generating a minimum of false positives. Sensitivity to mM binding is important as small fragments may still exhibit good LE despite a KD of 2 to 3 mM, and certain target classes, such as protein-protein interaction targets, 24 inherently bind fragments more weakly. Moreover, even if a fragment binds with KD in the range of 10 mM, we have readily found close analogs with KD 20-fold tighter, which could result in an increase of the LE by 0.08 to 0.1. Below we discuss each of the 4 methods in light of these characteristics.
Ligand-observed NMR and STD, as first proposed by Mayer and Meyer, 11 and a similar method known as waterLOGSY 25 are widely used NMR techniques for detecting fragment binding. STD and waterLOGSY have the advantage of being relatively easy to implement, requiring only standard NMR instrumentation (optimally with a cryoprobe head) and alleviating the need for an isotopically labeled target, while reducing the amount of protein required by a factor of 10. On the other hand, the sensitivity of STD is dependent on the efficiency of saturation transfer, which is a function of the correlation time τc of the target. Because τc generally scales with the size of a molecule, both STD and waterLOGSY become more efficient with increasing molecular weight of the target protein, and therefore neither method is optimal for proteins less than about 20 kDa. Furthermore, the protein must remain soluble even in the presence of high concentrations of fragments in low salt (less than 50 mM total ionic strength) to take advantage of the increased sensitivity afforded by a cryoprobe. If the latest generation cryoprobe with a sample volume of 30 µL is used, then 1500 compounds can be screened using about 15 mg of a target the size of RdRP. However, more typically, a 5-mm probe is available, requiring a reduction of the protein concentration of 5 µM, requiring about 50 mg of protein. In this case, the real limitation of solution ligand-observed NMR methods is the lack of sensitivity to weak binding in the mM range.
The use of TINS as a primary screening technique has a number of potential advantages. Our results clearly indicate that it is most sensitive to weak binding. For example, in one project, a ligand with a KD of 8.9 mM was found that, due to Mr 135 Da, had a LE of 0.28 (to be published elsewhere). Subsequently, a crystal structure of the target-fragment complex was solved, enabling rational elaboration. TINS also makes efficient use of the target, requiring only 4 mg of RdRP to screen the entire fragment library while ligand binding and enzymatic activity of targets routinely is unchanged even after screening 1500 fragments. 26 The use of a reference reduces the number of false positives and, by careful selection of the reference, allows one to adjust the scope of a screen to, for example, focus on one binding site or select fragments specific for one member of a protein family. Using the PH domain of human Akt1, we routinely observe 7 to 10 compounds per screen that preferentially bind the reference, as compared to 30 to 100 that preferentially bind the target. Last, TINS is applicable to a wide variety of targets, more than 20 different proteins, including membrane proteins that have been screened.
We noted a distinct lack of correlation between the binding signal for TINS and the potency in the biological activity, whereas STD fares only marginally better. The poor correlation between the level of biological activity and the magnitude of the NMR readout is likely due to the dependence of ligand-observed NMR signals on the off rate of the ligand. During the fixed length of an NMR experiment, tighter binding ligands, which have slower off rates, will not generate as much signal amplification as weaker binding ligands. This is the likely explanation for the observation that the 2 fragments with slow kinetics in SPR and potent biological activity were only marginally detected as hits in both TINS and SPR. A further issue for TINS is that for ligand screening, the parameters of the NMR experiment are optimized to detect weak binders. By shortening the T2 period during the spatially selective TINS experiment, the correlation between TINS effect and binding affinity can be improved considerably. 27 However, because TINS depends on enhanced transverse relaxation of ligands in the bound state to generate the binding signal, significant inherent differences in R2 between fragments render precise quantitation difficult.
Because the ultimate goal of a small-molecule drug is to modulate the biological activity of a target, using such an assay in the initial stage of drug discovery is logical. This approach has the advantage that it is inherently quantitative. However, high-concentration screening is not a generally applicable approach. First, not all targets have a biological activity than can be readily assayed in vitro, and cell-based assays do not typically possess sufficient sensitivity to detect weakly active fragments. Even when an efficient in vitro biological assay can be developed, it may not be appropriate for screening fragments. For example, protein-protein interactions can often be readily measured using a fluorescently labeled binding partner. However, the low nM affinities characteristic of these interactions are often too tight to be displaced by fragments that bind in the mM range. Last, the high concentration of fragments necessary to generate a response sufficiently above the noise level in a biological assay may lead to artifactual mechanisms of inhibition such as aggregation. 28 However, the combination of a biological assay with a high-quality biophysical assay allows one to eliminate false positives that act through aggregation or nonspecific destabilization mechanisms. Thus, we view biochemical assays, when possible, as an ideal hit validation and prioritization tool after a biophysical assay.
Calculations based on the fundamental principles of SPR suggest that for a ligand of 150 Da, the concentration of the ligand in the SPR assay must be between 0.5 and 1.2 times the KD for targets in the typical molecular mass range of FBDD. 29 It is unusual to screen fragments for binding at concentrations higher than 500 µM9 in an SPR assay, which places an approximate upper limit of detection of 1 mM KD for moderate-sized targets. These calculations are borne out by our experiments where 82% of the group I fragments, which have sub-mM KDs, were detected by SPR, but only a small number of group II fragments, which include many compounds with KD > 1 mM, could be detected. Therefore, our data suggest that given the present sensitivity of SPR instruments, use of this method as a primary screening approach may lead to an unacceptably high level of false negatives. However, the highly quantitative nature of SPR makes this an extremely powerful, orthogonal technique to validate and prioritize hits from the primary screen. In particular, the combination of TINS for primary fragment screening, in which the format lends itself to ready confirmation of biological activity, and SPR for validation/prioritization should prove a powerful combination to rapidly find and evaluate weakly binding fragment hits for structural studies and elaboration projects.