
Abstract
The p53 tumor suppressor protein plays a pivotal role in suppressing oncogenesis by regulating a range of cellular functions, including DNA repair, cell growth, cell cycle progression, and cellular death. A network of different pathways converge upon p53, ultimately regulating the response of the tumor suppressor protein by posttranslational modifications. The authors have developed a time-resolved fluorescence resonance energy transfer (TR-FRET)–based high-throughput compatible assay to analyze the critical posttranslational modifications of p53, including phosphorylation, acetylation, and ubiquitination. By using full-length p53 protein fused with GFP (GFP-p53) as the substrate, they were able to measure all 3 different posttranslational modifications with a single substrate. In addition, with a few additional steps, the GFP-p53 substrate can also be used to assay deacetylation to aid in the discovery of inhibitors for sirtuins or other deacetylase enzymes. The flexibility of the assay to measure a diverse range of posttranslational modifications allows one to further dissect the complex regulating mechanisms of p53 and enable the discovery of specific inhibitors for these processes.
Introduction
T
The intricate regulation of p53 by posttranslational modification has been extensively studied, and a number of excellent review articles are available on this topic. 1,4-6 Historically, phosphorylation has been viewed as the major mechanism of protein regulation within cellular pathways and has therefore been a major focus of drug discovery programs. p53 is regu lated by phosphorylation by a number of DNA damage kinases such as ATM, ATR, and DNA-PK ( Fig. 1 ). Recently, other posttranslational modifications, including acetylation and ubiquitination, have been identified to play key regulatory steps in the activation and stability of p53. Unfortunately, the majority of drug discovery tools available to study posttranslational modifications of p53 have been limited to either low- to medium-throughput assays methods such as Western blots or enzyme-linked immunosorbent assays (ELISAs) or high-throughput methods that use substrate mimics or small peptides centered on the site of modification. 7,8 Although these small peptide substrates are good for studying a single posttranslational event such as phosphorylation, they lack the necessary secondary structure and binding domains that are required to study more complex regulatory interactions that require the full-length protein. In addition, the use of substrate mimics can sometimes lead to an incomplete or erroneous picture of the true biological function of the protein. 7
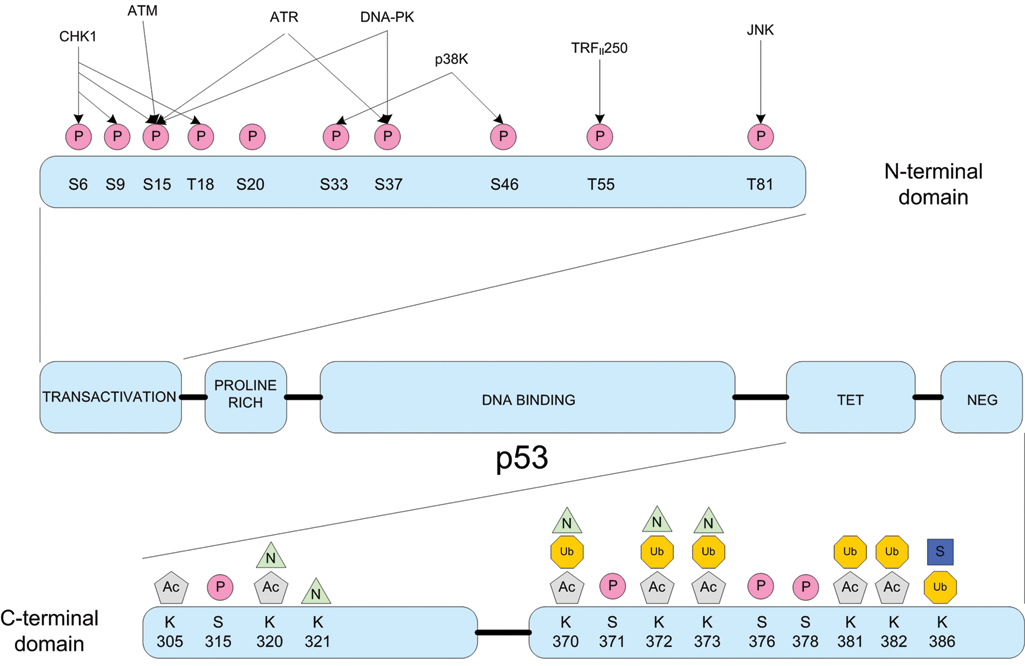
Posttranslational modifications of p53. A wide combination of posttranslational modifications regulates the tumor suppressor protein p53. Posttranslational events such as phosphorylation (P), acetylation (Ac), ubiquitination (Ub), sumoylation (S), and NEDDylation (N) all play pivotal roles in affecting p53 stability and activity. Adapted from Lavin and Gueven 5 and Olsson et al. 6
In an attempt to better represent the physiological system, we have developed an assay that uses the full-length p53 protein as the substrate. By expressing the p53 protein as a fusion with green fluorescent protein (GFP), a high-throughput time-resolved fluorescence resonance energy transfer (TR-FRET)–based assay can be developed to study a diverse range of posttranslational modifications of p53, including phosphorylation, acetylation, deacetylation, and ubiquitination. By using the full-length p53 as the substrate, all of the necessary sites of modifications and protein binding sites are present to facilitate the study of multiple posttranslational modifications. This not only helps streamline assay development due to the use of a single substrate for all modifications, but allows for maximal flexibility in the specific target or posttranslational modification to be studied.
Materials and Methods
Purified recombinant kinases, the LanthaScreen® amine reactive terbium chelate, and LanthaScreen® assay reagents for the phosphorylation and ubiquitination assays were all obtained from Invitrogen, part of Life Technologies (Carlsbad, CA). For the acetylation assay, recombinant p300 was obtained from Protein One (Bethesda, MD), and the acetyl-p53 (Lys382) antibody was obtained from Cell Signaling Technology (Danvers, MA). SIRT1, SIRT2, and SIRT4 were obtained from BPS Biosciences (San Diego, CA), whereas SIRT3 was obtained from Enzo Life Sciences (Plymouth Meeting, PA). Ubiquitin enzymes (E1 and Hdm2) were obtained from Boston Biochem (Cambridge, MA), and the E2 (UbcH5a) was obtained from Enzo Life Sciences. General chemicals and small-molecule inhibitors were obtained from Sigma-Aldrich (St. Louis, MO) or EMD Biosciences (San Diego, CA).
All assays were performed in 384-well low-volume assay plates (Corning No. 3676), with a final volume of 20 µL. For the dose-response curves, a GFP-only control experiment was performed in parallel to determine what portion of the assay signal (if any) was due to the posttranslational modification of the GFP portion of the substrate. In all cases, the GFP-only control did not contribute to the assay signal, suggesting that the change in signal during the assay was due to modification of p53 and not that of GFP (see supplemental information).
Expression and purification of GFP-p53
An expression plasmid encoding a his-tagged GFP-p53 (NP_00537; P278A) fusion was expressed and purified from baculovirus using standard methods. Briefly, recombinant baculovirus was generated using the Bac-to-Bac Baculovirus Expression System following the manufacturer’s recommendation (Invitrogen). The full-length GFP-p53 protein was expressed in Sf9 insect cells transfected with baculovirus at a multiplicity of infection (MOI) of 10. The cells were harvested by centrifugation and then resuspended and lysed in break buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 25 mM imidazole, 0.1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride [PMSF], 5 mM benzamidine, 20 µg/mL leupeptin, 10µM E-64, 1× phosphatase inhibitor cocktail set III [CalBiochem, San Diego, CA], and 1× phosphatase inhibitor cocktail 1 [Sigma-Aldrich]) at a ratio of 10 mL of break buffer per gram of cell paste. The cells were further disrupted with the use of a handheld biohomogenizer and then sonicated on ice. Cellular debris was removed by centrifugation at 10,000 g for 60 min at 4 °C. The recovered supernatant was incubated with 1 mL of Ni-NTA (nickel nitrilotriacetic acid) per gram of cell paste. The Ni-NTA was recovered by centrifugation and packed into a disposable column, where it was washed with 10 bed volumes (Bv) of Ni-NTA wash buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, and 30 mM imidazole) followed by a bump elution of 4 (Bv) of Ni-NTA elution buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 400 mM imidazole, 10% glycerol). The protein was then dialyzed into storage buffer (10 mM HEPES, 245 mM NaCl, 4.8 mM KCl, and 10% glycerol) and stored at –80 °C until use. The yield was 0.9 mg/g cell paste of 90% purity.
Labeling of antibody
The standard labeling protocol supplied with the amine reactive terbium chelate was used to label carrier-free acetyl-p53 (Lys382) antibody. Briefly, the amine-reactive terbium chelate was dissolved to a concentration of 2.5 mg/mL in 1.0 M sodium bicarbonate, pH 9.4. The dissolved chelate was then immediately added to the antibody at a 1:10 (vol/vol) ratio and allowed to incubate at room temperature for 4 h. Following the labeling reaction, the antibody was dialyzed twice against HEPES-buffered saline (HBS) to remove unreacted and hydrolyzed label. The labeling efficiency (chelates per antibody) was calculated from the absorbance of the chelate at 343 nm (ϵ343 = 12,570 M−1 cm−1) and the absorbance of the antibody at 280 nm (ϵ280 = 210,000 M−1 cm−1) after subtracting out the absorbance of the chelate at 280 nm (1.1 times its absorbance at 343 nm). Under these conditions, a labeling efficiency of 4 to 9 chelates per antibody was typical.
Phosphorylation assay
Kinase reactions were performed in a final volume of 10 µL at room temperature in an assay buffer consisting of kinase buffer A (PV3189; 50 mM HEPES [pH 7.5], 0.01% BRIJ-35, 10 mM MgCl2, and 1 mM EGTA) supplemented with 1 mM dithiothreitol (DTT) and 2.5 µg/mL of sheared DNA. For the kinase titration, a dilution series of DNA-PK was performed with 200 nM GFP-p53 and 10 µM adenosine triphosphate (ATP). After 1 h, a 10-µL solution of terbium-labeled anti-phospho-p53 (pSer15) antibody (PV5128) and EDTA prepared in TR-FRET dilution buffer (PV3574) was added to each well, for a final concentration of 2 nM antibody and 10 mM EDTA. After a 1-h equilibration period, the plate was read on a TR-FRET-compatible plate reader, and a response ratio was calculated by dividing the emission ratio (520 nm/495 nm) of the assay signal by the emission ratio of a background well. For the inhibitor titrations, a fixed concentration of DNA-PK (204 ng/mL) was combined with 200 nM GFP-p53, 10 µM ATP, and a dilution series of inhibitor. Following the 1-h incubation period, the terbium-labeled antibody and EDTA were added as outlined above.
Acetylation assay
The acetylation reactions were performed in a final volume of 10 µL at 30 °C in an assay buffer consisting of 50 mM Tris (pH 8.0), 10% glycerol, 0.1 mM EDTA, 1 mM DTT, and 10 µM acetyl-CoA. For the acetylation assay, a dilution series of p300 was combined with 200 nM GFP-p53 and 10 µM ATP in assay buffer. After 1 h, a 10-µL solution of terbium-labeled anti-acetyl-p53 (Lys 382) prepared in TR-FRET dilution buffer was added to each well, for a final concentration of 2 nM antibody. After a 1-h equilibration period, the plate was read on a TR-FRET-compatible plate reader, and a response ratio was calculated. For the inhibitor titration, a fixed concentration of p300 (3.5 ng/mL) was combined with 200 nM GFP-p53, 10 µM ATP, and a dilution series of inhibitor curcumin. Following the 1-h incubation period, the terbium-labeled antibody was added as outlined above.
Deacetylation assay
A bulk preparation of acetylated GFP-p53 was performed as outlined in the acetylation assay protocol above, except the volume and amount of proteins in the reaction were scaled up appropriately. A 1:6 ratio (wt/wt) of p300 to GFP-p53 was used and incubated at 30 °C for 8 h. Following the acetylation reaction, the acetylated GFP-p53 was purified using the 6x His affinity tag on the GFP-p53 and a Ni-NTA column using standard chromatography techniques.
For the deacetylation reactions, a dilution series of SIRT1 to SIRT4 was performed against 50 nM acetyled GFP-p53 and 1 mM nicotinamide adenine dinucleotide (NAD) at 30 °C in assay buffer consisting of 50 mM Tris (pH 8.0), 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, and 1 mM DTT. After 1 h, a solution of terbium-labeled anti-acetyl-p53 (Lys 382) in TR-FRET dilution buffer was added to each well to obtain a final concentration of 2 nM. Following a 1-h equilibration period, the plate was read on a TR-FRET-compatible plate reader, and the percent deacetylation (compared to a control) was calculated. For the inhibitor titration, a fixed concentration of SIRT1 (0.5 µM) was combined with 50 nM acetylated GFP-p53, 1 mM NAD, and a dilution series of inhibitor. The same method was followed with the resveratrol experiments except the concentrations of SIRT1 and SIRT2 were 23.7 nM and 47.6 nM, respectively. Following the 1-h incubation period, the terbium-labeled antibody was added as outlined above.
Ubiquitination assay
The ubiquitination reactions were performed in a final volume of 10 µL at room temperature in an assay buffer consisting of 100 mM Tris (pH 8.0), 1 mM DTT, and 10 mM MgCl2. A dilution series of E3 (Hdm2) was combined with 50 nM GFP-p53, 10 nM E1, 100 nM E2 (UbcH5a), 50 nM terbium-ubiquitin (PV4376), and 1 mM ATP. After 1 h, 10 µL of TR-FRET dilution buffer was added to each well, and the plate was then read on a TR-FRET-compatible plate reader and a response ratio was calculated. For the inhibitor titrations, a fixed concentration of Hdm2 (50 nM) was combined with 50 nM GFP-p53, 10 nM E1, 100 nM E2 (UbcH5a), 50 nM terbium-ubiquitin, 1 mM ATP, and a dilution series of inhibitor. Following the 1-h incubation period, TR-FRET dilution buffer was added as outlined above.
Results and Discussion
Assay principle
The basic principle of using physiological GFP substrate fusions in the development of a TR-FRET assay has been demonstrated previously. 9-11 Although typically, these assays have focused on phosphorylation, the same technology can also be used to study a broader range of posttranslational modifications, including acetylation, deacetylation, and ubiquitination, to name a few. The ability to study multiple posttranslational modifications on a single protein is key to understanding the complex regulation of gateway proteins such as p53.
The basic assay schematics for the posttranslational modifications of p53 are displayed in Figure 2 . The GFP-p53 substrate is combined with the necessary enzyme and cofactors to perform the posttranslational modification of interest. Following the modification of the GFP-p53 substrate, a terbium-labeled antibody directed toward the site of modification is added to bind to the modified substrate. Upon binding of the antibody to the GFP-p53 substrate, FRET can occur between the FRET donor (the LanthaScreen® terbium chelate on the antibody) and the FRET acceptor (the GFP on the substrate) and result in an increase in fluorescence signal at 520 nm. Because of the inherent properties of FRET, signal is observed only when the FRET donor is in close proximity to the FRET acceptor on the substrate. Although other posttranslational modifications may occur within the reaction, only the modification that the site-specific antibody is directed toward results in a FRET signal. In the absence of the posttranslational modification, the antibody remains unbound and FRET does not occur. Additional posttranslational modification assays for p53 (such as acetylation) can be performed easily by simply altering the terbium-labeled site-specific antibody that is added to the reaction mixture.
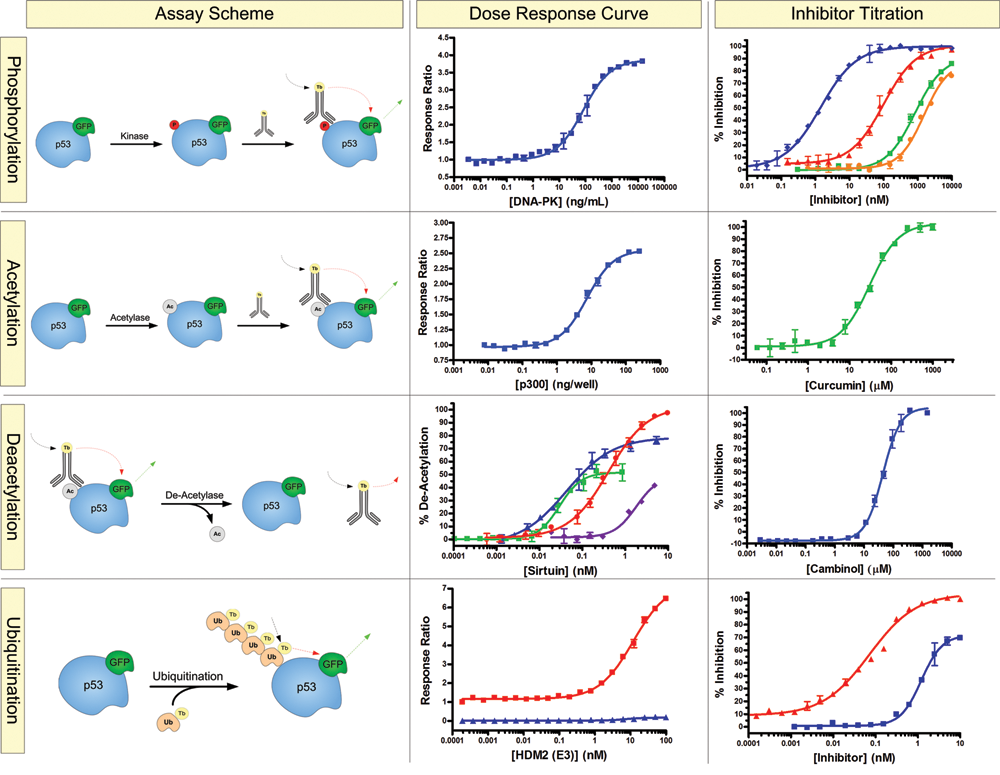
Biochemical posttranslational modification assays for p53. Phosphorylation: An assay scheme outlining the biochemical assay for p53 phosphorylation. A dose-dependent phosphorylation of the GFP-p53 substrate by the protein kinase DNA-PK (Z′ = 0.94), followed by inhibition with PI-103 (♦; Z′ = 0.86), Compound 401 (▲; Z′ = 0.89), 1-(2-hydroxy-4-morpholin-4-yl-phenyl)ethanone (■; Z′ = 0.91), and Aryl-Morpholine Analog 37 (●; Z′ = 0.82). Acetylation: An assay scheme outlining the biochemical assay for p53 acetylation. A dose-dependent acetylation by p300 is shown (Z′ = 0.83), followed by inhibition with the p300 inhibitor, curcumin (Z′ = 0.88). Deacetylation: An assay scheme outlining p53 deacetylation. A dose-dependent deacetylation of acetyl-GFP-p53 by SIRT1 (■; Z′ = 0.71), SIRT2 (▲; Z′ = 0.96), SIRT3 (●; Z′ = 0.75), and SIRT4 (♦), followed by inhibition of the deacetylation reaction of SIRT1 with cambinol (Z′ = 0.84). Ubiquitination: An assay scheme outlining p53 poly-ubiquitination with Hdm2 in the presence (■) or absence (▲) of adenosine triphosphate (Z′ = 0.85). This is followed by inhibition of the ubiquitination reaction with RITA (■; Z′ = 0.86) and (–) nutlin (▲; Z′ = 0.93).
Phosphorylation of p53
Regulation of p53 by phosphorylation occurs primarily in the N-terminal transactivation domain of p53 (see Fig. 1 ). Physiological stresses, such as UV light and γ-irradiation that can cause DNA damage, stimulate the activity of the DNA damage kinases, including ATM or DNA-PK, to phosphorylate p53 at multiple sites, including serine 15. Although the exact consequences of phosphorylation of p53 at serine 15 are still under investigation, modification at this site is thought to stimulate further posttranslational modifications of p53, including interactions with transcriptional activators such as p300. 3 A dose-dependent phosphorylation of GFP-p53 at serine 15 was observed upon the addition of DNA-PK to the reaction mixture ( Fig. 2 ). Inhibition of the phosphorylation event could also be observed upon the addition of known DNA-PK inhibitors. A strong correlation of IC50 values was observed with known literature values (see supplemental information) and when the same assay was performed with the more traditional peptide-based substrate surrounding serine 15 of p53 (data not shown).
Acetylation/deacetylation of p53
As with phosphorylation, the same physiological stresses that damage DNA can also stimulate acetylation of p53 ( Fig. 1 ). Acetylation of p53 helps stimulates DNA binding and consequently transcriptional activation of target genes. 6 In addition, acetylation also appears to have a regulatory role in the ubiquitination and subsequent proteasomal degradation of p53. 6
To measure the acetyl transferase activity of p300 on GFP-p53, a site-specific antibody directed toward acetyl–lysine 382 of p53 was used. A dose-dependent acetylation of GFP-p53 was observed with increasing concentrations of p300 ( Fig. 2 ). To further validate the acetyl transferase activity of p300, a titration of curcumin, a known inhibitor for p300 activity, resulted in an IC50 value that was consistent with known literature values ( Fig. 2 ).
The same assay system can also be used to measure deacetylase activity such as that of sirtuins or other related histone deacetylase (HDAC) enzymes. The deacetylation counteracts (or downregulates) the transcriptional activation of p53 and suppresses the apoptotic activity of the tumor protein. 4,6 It is this central role of the HDACs (more specifically the sirtuins) in the regulation of the transcriptional and apoptotic function of p53 that make them interesting drug candidates and a focus for drug discovery programs. The principle of the deacetylation assay is similar to the acetylase assay, except that a decrease in FRET signal is obtained upon the removal of the acetyl group from p53 ( Fig. 2 ). An epitope tag on the GFP-p53 allows the substrate to be quickly purified from the reaction mixture following acetylation by p300. It is this purified acetylated GFP-p53 that becomes the starting substrate for the deacetylation assay.
The deacetylase activity of sirtuins 1-4 can be measured using the acetyl-GFP-p53 substrate ( Fig. 2 ). Previous studies of SIRT4 using a substrate mimic failed to show any deacetylase activity even though SIRT4 shares a conserved catalytic domain with the remaining sirtuins. 12,13 However, using the GFP-p53 full-length substrate, deacetylase activity of p53 by SIRT4 was reproducibly observed using the same assay conditions as those of the other sirtuins, although considerably less deacetylase activity was observed than that of SIRT1 to SIRT3 ( Fig. 2 ). Inhibition of SIRT1 deacetylase activity with cambinol ( Fig. 2 ) as well as activation of both SIRT1 and SIRT2 using resveratrol ( Fig. 3 ) was performed with a strong correlation to known literature values.
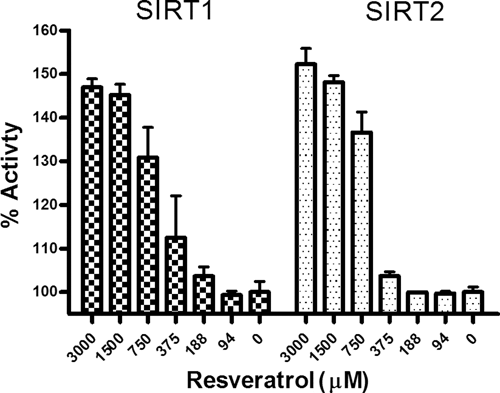
Activation of SIRT1 and SIRT2 with resveratrol. The activation of sirtuins with resveratrol has been a controversial topic. Some fluorescence-based assays using a peptide-based substrate mimic have shown significant activation of SIRT1 with micromolar concentrations of resveratrol. 7 However, using the full-length GFP-p53 substrate, no increase in activity of SIRT1 or SIRT2 is observed until much higher mM concentrations of resveratrol were achieved.
Ubiquitination of p53
To help maintain tight regulation over cellular levels of p53, an intricate balance between the ubiquitination ligase Hdm2 and p53 is continuously in flux. The ubiqutin ligase Hdm2 is responsible for the poly-ubiquitination and subsequent proteasomal degradation of p53. Cellular pools of p53 are continuously being recycled under the watchful regulation of Hdm2 to keep the apoptotic role of p53 at check. Biochemical assays for the identification of inhibitors for the poly-ubiquitination of p53 are of interest to cancer researchers who want to interfere with the Hdm2-p53 interaction and induce apoptosis in carcinogenic cells.
Unlike other posttranslational modifications such as phosphorylation and acetylation, which involve the transfer of a small chemical entity, ubiquitin in comparison is a large protein that can be directly modified with the FRET donor (i.e., the terbium chelate), eliminating the need for the use of antibodies in the assay ( Fig. 2 ). During the ubiquitination reaction, GFP-p53 is modified with a terbium-labeled ubiquitin via an iso-peptide bond between the C-terminal glycine of ubiquitin and a lysine of p53. The ubiquitination event brings the FRET donor into close proximity to the FRET acceptor on the substrate (i.e., GFP), resulting in an increase in FRET signal.
A dose-dependent increase in assay signal is observed with increasing concentration of Hdm2 ( Fig. 2 ). Elimination of ATP or any of the necessary ubiquitination enzymes (i.e., activating enzyme [E1] or conjugating enzyme [E2]) results in the failure of a FRET signal to develop. Compounds known to interrupt the Hdm2-p53 binding interaction (RITA and nutlin) inhibited the ubiquitination reaction and showed good agreement with literature values, providing further validation of the assay format. Modification of p53 with other ubiquitin-like proteins such as SUMO and NEDD8 (data not shown) adds an additional level of complexity and regulation to an already diverse range of posttranslational modifications.
The regulation of p53 is controlled by a complex combination of posttranslational modifications, including phosphorylation, acetylation, and ubiquitination. Although individually, each posttranslational modification plays an important step in the regulation or stability of p53, it is the interplay of multiple different posttranslational modifications that ultimately dictates the biological role and outcome of this protein.
By using a GFP fusion of the full-length p53 as the substrate, we developed high-throughput TR-FRET-based assays to analyze critical posttranslational modifications of p53. With the use of the single GFP-p53 substrate, multiple assays can be quickly developed by simply altering the terbium-labeled antibody (or terbium-labeled protein) directed toward the posttranslational modification of interest. In addition, by incorporating an affinity tag on the GFP-p53 substrate, one can create precharged substrates to study complex signaling pathways that require a premodified substrate to promote further sequential posttranslational modifications.