
Abstract
Macrophage migration inhibitory factor (MIF) is a major mediator of innate immunity and inflammation and presents a potential therapeutic target for various inflammatory, infectious, and autoimmune diseases, including cancer. Although a number of inhibitors have been identified and designed based on the modification of known nonphysiological substrates, the lack of a suitable high-throughput assay has hindered the screening of chemical libraries and the discovery of more diverse inhibitors. Herein the authors report the development and optimization of a robust high-throughput kinetic-based activity assay for the identification of new MIF inhibitors. Using this assay, they screened 80,000 small molecules and identified and validated 13 novel inhibitors of MIF catalytic activity. These small molecules demonstrated inhibition constant (Ki,app) values ranging from 0.5 to 13 µM.
Keywords
Introduction
M
MIF’s 3D structure was resolved in 1996. Subsequent biochemical and mutagenesis studies have allowed the identification of the key residues involved in forming the catalytic site and in regulating MIF’s tautomerase activity. 9 Since that time, significant efforts have attempted to identify small-molecule modulators of MIF by targeting its tautomerase activity. 10 The availability of X-ray structures of the MIF trimer and the tautomerase catalytic site has facilitated these efforts and has stimulated great interest in rationally developing structure-based inhibitors. The first MIF inhibitors were identified by testing different D-dopachrome derivatives or phenyl pyruvic acid analogs. 11 In the past decade, several classes of MIF inhibitors have been developed by introducing modifications on substrate analogs and by screening focused libraries of natural products. 10,12 Several of these inhibitors were later shown to modulate MIF’s biological activities (e.g., IS01 and OXIME11) in cellular models and in vivo. 13,14
The lack of a robust high-throughput activity assay has hindered the screening of large chemical libraries and the discovery of diverse MIF inhibitors, including allosteric inhibitors. Therefore, with the exception of this report, the screening of large chemical libraries has been performed only via molecular dynamics, docking, and other in silico computational approaches. 7,13,14 The instability of MIF substrates D-dopachrome methyl ester and phenyl pyruvic acid has been cited as the major obstacle in developing high-throughput screening (HTS) chemical assays for MIF. 10 In the absence of MIF, the D-dopachrome methyl ester is unstable and undergoes slow spontaneous decarboxylation to form a 5,6-dihydroxyindole (DHI) and CO2 at physiologic pH. 15 To circumvent this, the D-dopachrome substrate can be freshly prepared via chemical oxidation using excess periodate, which increases the rate of spontaneous reaction. The removal of periodate by reverse-phase chromatography has been shown to improve substrate stability. 16 Unlike D-dopachrome, phenyl pyruvic acid exhibits a slow rate of tautomerization to the enol form in acetate buffer (pH 6). Accordingly, the substrate stock solutions must equilibrate for at least 12 h to avoid any increase in background absorbance by residual enols and should be kept away from oxygen. 17
With a stable substrate in hand, we sought to develop a suitable assay for HTS to facilitate the screening of large diverse chemical libraries and to identify novel competitive inhibitors. Because the linear range of the reaction was too short for an endpoint assay, a kinetic assay was performed instead, which continuously monitors enzymatic activity.
Enzymatic assays are generally developed as endpoint assays in HTS to simplify both data analysis and assay steps. However, the main advantage of a kinetic assay is avoiding the interference of compound absorbance because the readout is the initial velocity, not the real absorbance, especially in our case, where the substrate absorbs at 300 nm. On the other hand, the target in an endpoint assay is the detection of inhibitors, rather than analysis of enzyme kinetics or the details of inhibition. Although it has been reported that endpoint progressive curve data measurements can be used to identify competitive inhibitors, the exact time of measurement needs to be defined for every enzyme. These measurements should take place at high substrate conversion levels to maximize the detection of competitive inhibitors. 18 However, kinetic readings rely on initial velocity measurements under conditions where the substrate conversion level is low. This is more informative as it leads to the identification of various classes of enzymatic inhibitors and may facilitate enzyme kinetic analysis. In this article, we report the development and application of an optimized kinetic-based assay to screen a library of 80,000 compounds with the aim of identifying the broadest variety of mechanistic classes of inhibitors. We identified and validated 13 novel inhibitors of MIF’s catalytic activity with inhibition constant values (Ki,app) ranging from 0.5 to 13 µM.
Materials and Methods
Reagent, enzyme, and substrates
The following reagents were purchased from Sigma Aldrich (St. Louis, MO): boric acid, sodium phosphate, sodium acetate, sodium meta-periodate, potassium phosphate, EDTA, hydroxyphenyl pyruvic acid (HPP), phenyl pyruvic acid (PP), and D-dopachrome methyl ester.
MIF expression and purification were preformed as previously described. 19,20
Equipment: Beckman Biomek NX (Beckman Coulter, Brea, CA), Multidrop Thermo Fisher (Waltham, MA), SpectraMax plus 384 (Molecular Devices, Sunnyvale, CA), Molecular Devices (monochromator), and EnVision (filter based; PerkinElmer, Waltham, MA).
MIF tautomerase assay adaptation to 384-well format
D-dopachrome methyl ester assay
The keto-enol tautomeric conversion of D-dopachrome methyl ester by MIF was carried out as described previously. 21 Briefly, a fresh stock solution of L-dopachrome methyl ester was prepared by oxidation of L3-4 dihydroxy phenylalanine methyl ester with sodium meta-periodate. To a 4-mM solution of L3-4 dihydroxyphenylalanine methyl ester in 5 mL of distilled water was added sodium meta-periodate to a final concentration of 8 mM. The solution was mixed and incubated for 5 min at room temperature (protected from light and kept in ice). The tautomerase enzymatic activity was measured in 50 mM potassium phosphate buffer with 0.5 mM EDTA at pH 6. MIF (100 nM) was added to 384-well plates to a 60-µL final reaction volume, and the decrease in absorbance at 475 nm was followed using a SpectraMax plus 384 spectrophotometer.
To enhance substrate stability, excess sodium meta-periodate was removed based on the procedures described by Bendrat et al 16 using C8 disposable columns (Waters, Milford, MA). Cartridges were activated with 5 column volumes of methanol and then equilibrated with 10 column volumes of distilled water. The substrate was then loaded at a flow rate of 0.5 mL/min, and unbound material was washed with 10 column volumes of water. Finally, the substrate was eluted with 100% methanol and stored at −80 °C until use.
PP and HPP
HPP or PP tautomerization by MIF in 0.5 M borate buffer was performed as previously described. 5,17,22 Hydroxyphenyl pyruvic acid (100 mM) was dissolved in 50 mM sodium acetate buffer (pH 6), left overnight to stabilize, and then stored at 4 °C before use. HPP is stable for up to 4 to 5 days at 4 °C; beyond that time, a decrease in maximal absorbance is observed. Both enzyme (100 nM) and HPP (2 mM) were added to the reaction buffer (0.5 M boric acid in 0.2 M sodium phosphate, pH 6.2). The increase in absorbance due to the formation of the HPP enol-borate complex followed at 300 nm. This reaction was scaled down from the cuvette format (1 mL working volume) to a 384-well plate format (60 µL final reaction volume), where 20 µL of enzyme was added to the well containing 20 µL of buffer. Finally, 20 µL of substrate was dispensed, and the absorbance was monitored at 300 nm. The phenyl pyruvic acid stock solution was prepared to 100 mM in the reaction buffer, and the same protocol was used for HPP.
Compound library
The compound library consisted of approximately 80,000 small molecules and includes compounds approved by the Food and Drug Administration (FDA), a purified natural products library, and compounds purchased from Peakdale (High Peak, UK), Maybridge plc. (Cornwall, UK), Cerep (Paris, France), Bionet Research Ltd. (Cornwall, UK), Prestwick (Ilkirch, France), Specs and Biospecs (CP Rijswijk, the Netherlands), ENAMINE (Kiev, Ukraine), Life Chemicals (Burlington, Canada), MicroSource Diversity System’s NINDS custom collection (Gaylordsville, CT), ChemBridge (San Diego, CA), and ChemDiv (San Diego, CA), as well as from various academic institutions. Compounds were selected from the different vendors by applying a series of filters, including clogP and predicted solubility. Generally, all of the small molecules adhere to Lipinski’s rules (i.e., molecular weight <500, H-bond donors ≤5, H-bond acceptors ≤10, and logP <5), contain a low proportion of known toxicophores (i.e., Michael acceptors and alkylating agents) and unwanted functionalities (i.e., imines, thiols, and quaternary amines), and have been optimized for maximal molecular diversity.
HTS tautomerase assay parameters
The parameters used to screen for MIF tautomerase inhibitors, based on the format proposed by Inglese et al., 23 are summarized in Table 1 . Briefly, 25 µL of 1200 nM enzyme was preincubated with 0.5 µL of 1.67 mM compounds for 20 min at room temperature (RT). Then, 20 µL of the compound-enzyme mixture was transferred to each well in a 384 clear-well Greiner plate (Greiner Bio-One, Monroe, NC) containing 20 µL of reaction buffer (0.5 mM boric acid, pH 6.0, and 0.2 mM sodium phosphate). The reaction was initiated by the addition of 20 µL of 6 mM substrate HPP, and the absorbance at 300 nm was monitored continuously for up to 3 min. Background was carried out in the absence of MIF.
Summary Table of HTS Assay for the Discovery of MIF’s Tautomerase Activity Inhibitor
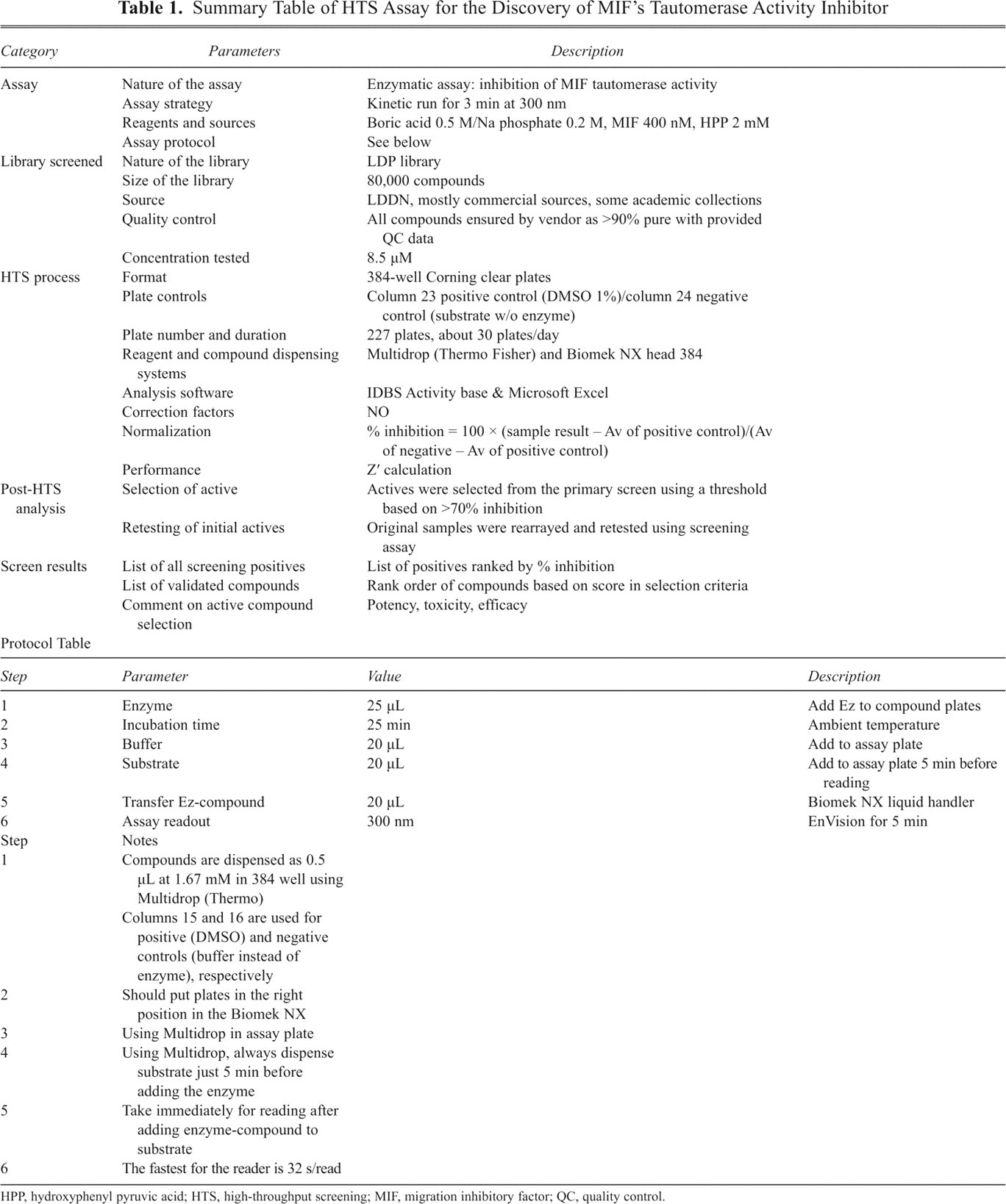
HPP, hydroxyphenyl pyruvic acid; HTS, high-throughput screening; MIF, migration inhibitory factor; QC, quality control.
Ki determination
MIF activity was tested at 12 different compound concentrations ranging from 0 to 50 µM. Promising compounds were initially dissolved to 10 mM in 100% DMSO and then serially diluted with 100% DMSO in 96-well plates. This was followed by a second dilution in reaction buffer and then transferred into a 384-well plate to yield a final DMSO concentration of 1.5%. Every concentration was tested in 4 replicates, and the assay was done in duplicate. Each compound-enzyme mixture was incubated for 25 min in buffer before the addition of substrate. The kinetics of the reaction was monitored at 300 nm for 3 min. Relative initial velocities were plotted as a function of inhibitor concentration. Data were fitted to the simple inhibition expression:
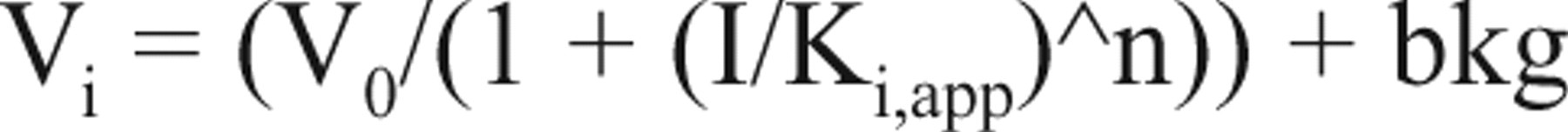
where V0 is the velocity at [I] = 0, and n is the Hill coefficient.
MALDI-TOF-MS MW measurements to examine the covalent modifier
Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS) was performed to determine if any of the hit compounds act via covalent modification of MIF. A mixture of 1 µL of sample (20 µM MIF incubated with 10 µM hit compound for 1 h) and 1 µL of sinapinic acid matrix solution was made, and then 1 µL of this mixture was deposited on top of the thin layer and allowed to air dry. The samples were analyzed with a 4700 MALDI-TOF/TOF instrument (Applied Biosystems, Foster City, CA).
Protein digestion analysis to identify the modified residue
In cases where the hits were shown to modify MIF, the modified protein was subjected to further analysis to identify the exact residue(s) being modified. The reaction mixture (10 µL of MIF sample previously incubated with 10 µM of covalent inhibitor for 1 h) was digested overnight with trypsin (Promega, Madison, WI). The digestion was stopped with 1 µL of formic acid and stored at 4 °C until further use. The samples were analyzed by MALDI-MS on a 4700 MALDI-TOF/TOF instrument (Applied Biosystems). Then, 1 µL of sample was mixed with 1 µL of DHB (20 mg/mL in 1% PA/ACN (1/1)), and 1 µL of this mixture was deposited on the target and allowed to air dry.
Compound toxicity studies
CellTiter 96 AQueous one-solution cell proliferation assay (Promega)
RAW 264.7 macrophages ATCC were plated at 50,000 cells/well with 100 µL final volume, and then the compounds were added at different concentration (0, 3, 10, and 20 µM) for an overnight incubation. For the cell proliferation assay, 20 µL of CellTiter 96 AQueous was added, and cells were incubated for 2 h at 37 °C. To measure the soluble formazan produced by cellular reduction of MTS, the absorbance was recorded at 490 nm.
CellTiter-Glo luminescent cell viability assay (Promega)
Compounds were added to the RAW 2647 macrophages as described above. After overnight incubation with the MIF inhibitors, plates were equilibrated at RT for 30 min, and then 100 µL of CellTiter-Glo was added to 100 µL of culture, and the plates were subjected to orbital shaking for 2 min to facilitate cell lysis. After a 10-min incubation at RT, the luminescent signal was measured using Tecan Sapphire II reader (Tecan, San Jose, CA).
Results
Development of the HTS assay
The goal of any screening program is to identify inhibitors with diverse inhibitory modalities. In enzymatic HTS assays, it is important to use substrate concentrations so that the major enzyme forms that accumulate in the steady state exist in near-equal concentrations. The initial velocity should yield a linear dependency on enzyme concentration, and the desired background should be less than 10% of the signal. We examined these parameters and optimized the assay for screening.
Selection of substrate
MIF functions as a tautomerase for 3 different substrates: D-dopachrome methyl ester, hydroxylphenyl pyruvic acid, and phenyl pyruvic acid ( Fig. 1A ). All 3 substrates were tested. The criteria for choosing the substrate for our assay were based on substrate stability, length of linear range, and ratio of signal to background. The D-dopachrome substrate did not meet these criteria. It is stable only for a maximum of 1 to 2 h when stored at 4 °C and even less at room temperature. Our efforts to improve the stability of this substrate were not successful. Removing excess sodium meta-periodate using a C8 reverse-phase disposable column did not provide reproducible results. Furthermore, changing the pH or ionic strength of the reaction buffer also did not result in improvements. The linear range of the kinetic assay for the D-dopachrome methyl ester is too short, at 40 s, and the signal-to-background ratio was too small, at less than 2-fold ( Fig. 1B ). Therefore, we sought to evaluate 2 other MIF substrates, HPP and PP. Both substrates yield a lower background and a longer linear range. PP, however, did not demonstrate tautomerase activity in disposable plastic cuvettes or in 384 clear-well plates (Greiner). For HPP, the formation of the enol-borate complex can be followed at 300 nm in disposable cuvettes and in 384-well plates. A linear range of 2 min was obtained with a signal to background greater than 2-fold ( Fig. 1C ). In addition, HPP in sodium acetate buffer (pH 6) is stable for several days at 4 °C, and the maximal absorbance is reproducible.
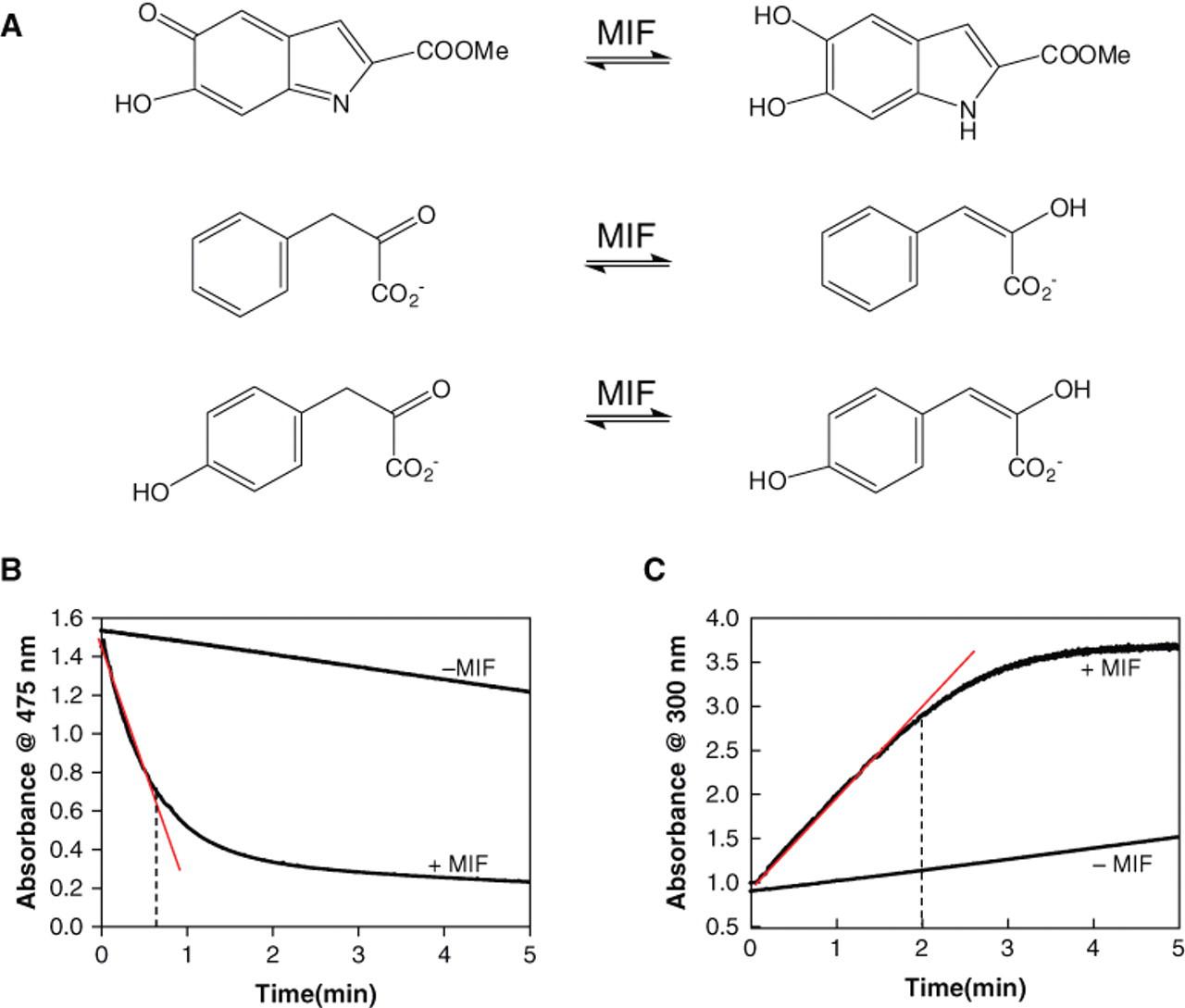
(
Assay optimization
For an enzyme assay, the first parameter that needs to be determined is the optimal substrate concentration to work with; this largely depends on the types of hits being sought. In general, concentrations near the Km value are optimal to achieve the widest range of enzyme inhibitor types, including competitive, uncompetitive, noncompetitive, and mixed inhibitors. Concentrations at the Km value allow the identification of inhibitors that interact with each possible state of the enzyme (free enzyme and enzyme-substrate complex in the reaction). Initial velocities were measured as a function of substrate concentration from 0 to 20 mM, and Km was determined by nonlinear least squares fit of the data to the simple Michaelis-Menten equation. The calculated Km average value is 2.1 ± 0.76 mM, which is in close agreement with the values reported in the literature: 2.4 mM 5 and 2.7 mM 22 . This value was used later in the screen and in the Ki,app calculation ( Fig. 2 ).
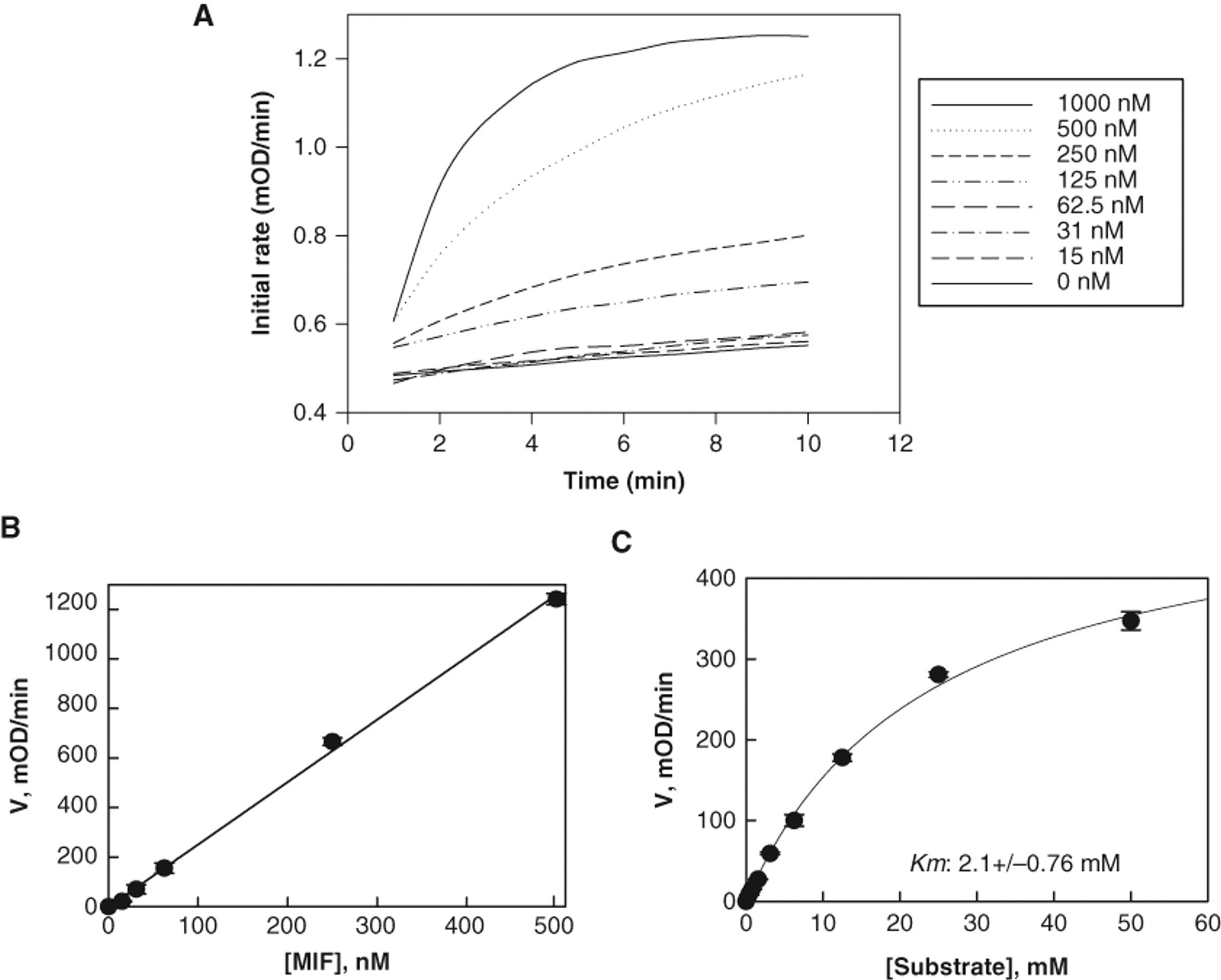
Optimization of the tautomerase assay reaction conditions: determining apparent Km of hydroxyphenyl pyruvic acid (HPP) and enzyme titration. (
Optimization of enzyme concentration
In enzymatic HTS assays, the concentration of enzyme should ensure the longest linearity with respect to enzyme stability. To determine the optimal concentration with respect to enzyme linearity response, we performed an enzyme dilution study and measured the respective initial velocity. Figure 2A shows MIF titration at an HPP concentration around the Km value. The Km was determined by measuring the initial rate as a function of substrate concentrations and the data fitted to the Michaelis-Menten equation V = Vmax*S/(S + Km). Up to 500 nM of MIF activity was linear. At higher enzyme concentrations, the substrate is depleted faster and linearity is lost.
Preincubation of MIF with compounds allows the identification of time-dependent inhibitors, including both irreversible and slow-binding inhibitors. To ensure that these types of inhibitors were not missed in the screening, enzyme-compound mixtures were incubated for 25 min prior to substrate addition. The experimental progress curve was then analyzed. An enzyme concentration of 100 nM gave a linear range of ~300 s with a signal window of 11%. Surprisingly, incubation of 100 nM MIF for 30 min at RT in 384-well plates resulted in a loss of MIF enzymatic activity by ~50%, which affected the signal background window. Increasing the concentration to 200 nM doubled the initial rate and provided an acceptable signal background window with a linear range of 200 s. In addition, the sensitivity was much lower when the detection system was read on the PerkinElmer EnVision plate reader, which is part of the fully automated robot system. This reader is filter based instead of monochromatic based. To address this, the enzyme concentration was increased to 400 nM for HTS to enhance the signal window ( Fig. 3 ).
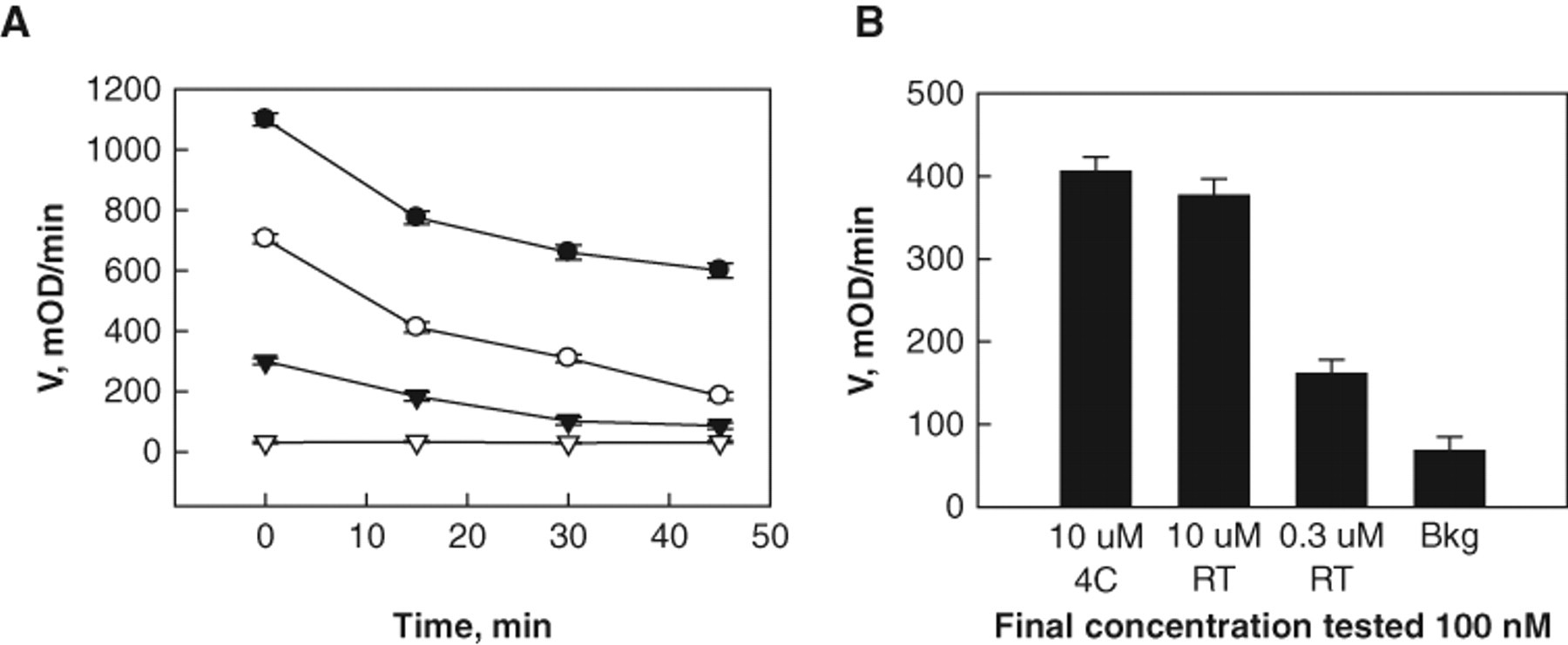
Determination of enzyme stability versus time. (
HTS screening
A total of 0.5 µL of 1.67 mM test compounds was included in single wells and assayed at a final concentration of 8.5 µM with a 0.66% final DMSO concentration. Sixteen wells containing 0.66% DMSO served as positive controls, and an additional 16 wells with substrate alone (without enzyme) served as negative controls. Compounds showing greater than 70% inhibition were defined as primary hits from the initial screen performed with 80,000 compounds. Representative data from 30,000 screened compounds are plotted ( Fig. 4 ). The Z′ values were calculated based on the inhibition of the enzyme initial rate during the first 3 min compared with positive and negative controls. 24 The average Z′ value of the screen was 0.6. The flowchart shown in Figure 5 summarizes the overall HTS procedure, including primary screens, data analysis, and Z′ calculation, followed by hit confirmation and validation. The Ki values were determined, and validated compounds were selected based on potency, toxicity, and chemical structure.
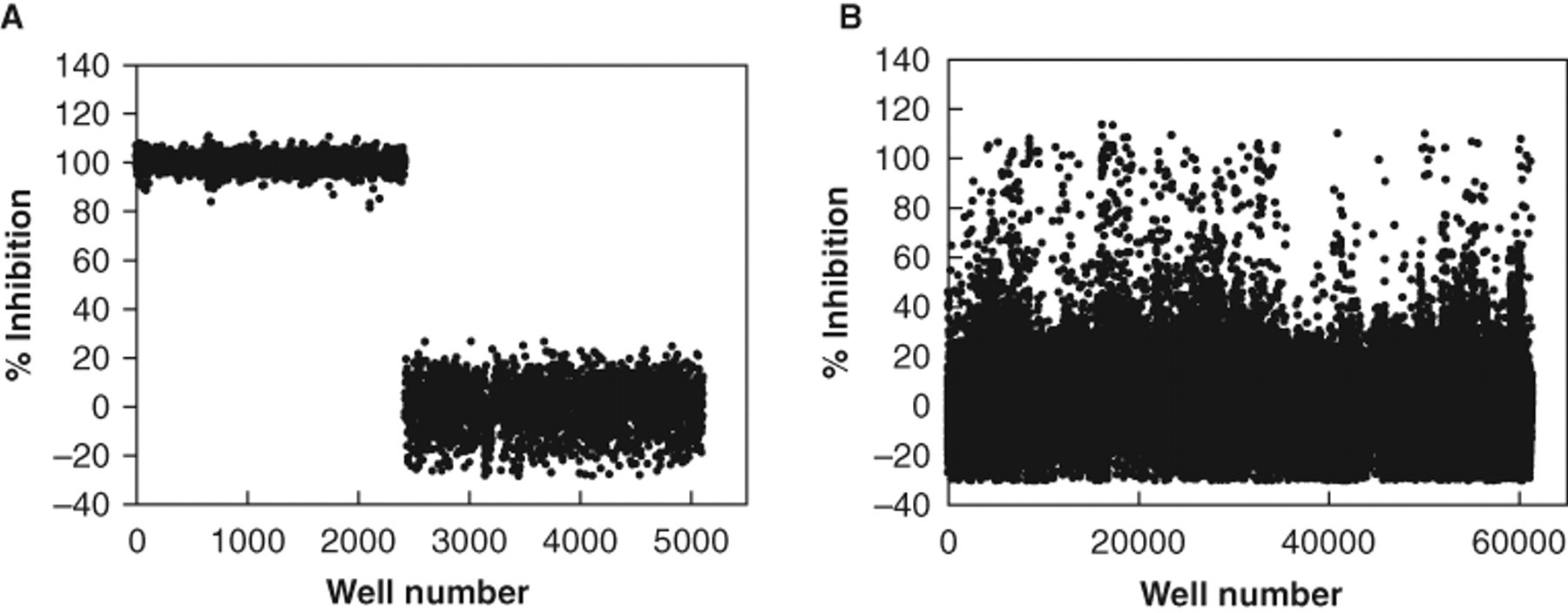
A graph of the high-throughput screening data from 80 representative 384-well plates. (
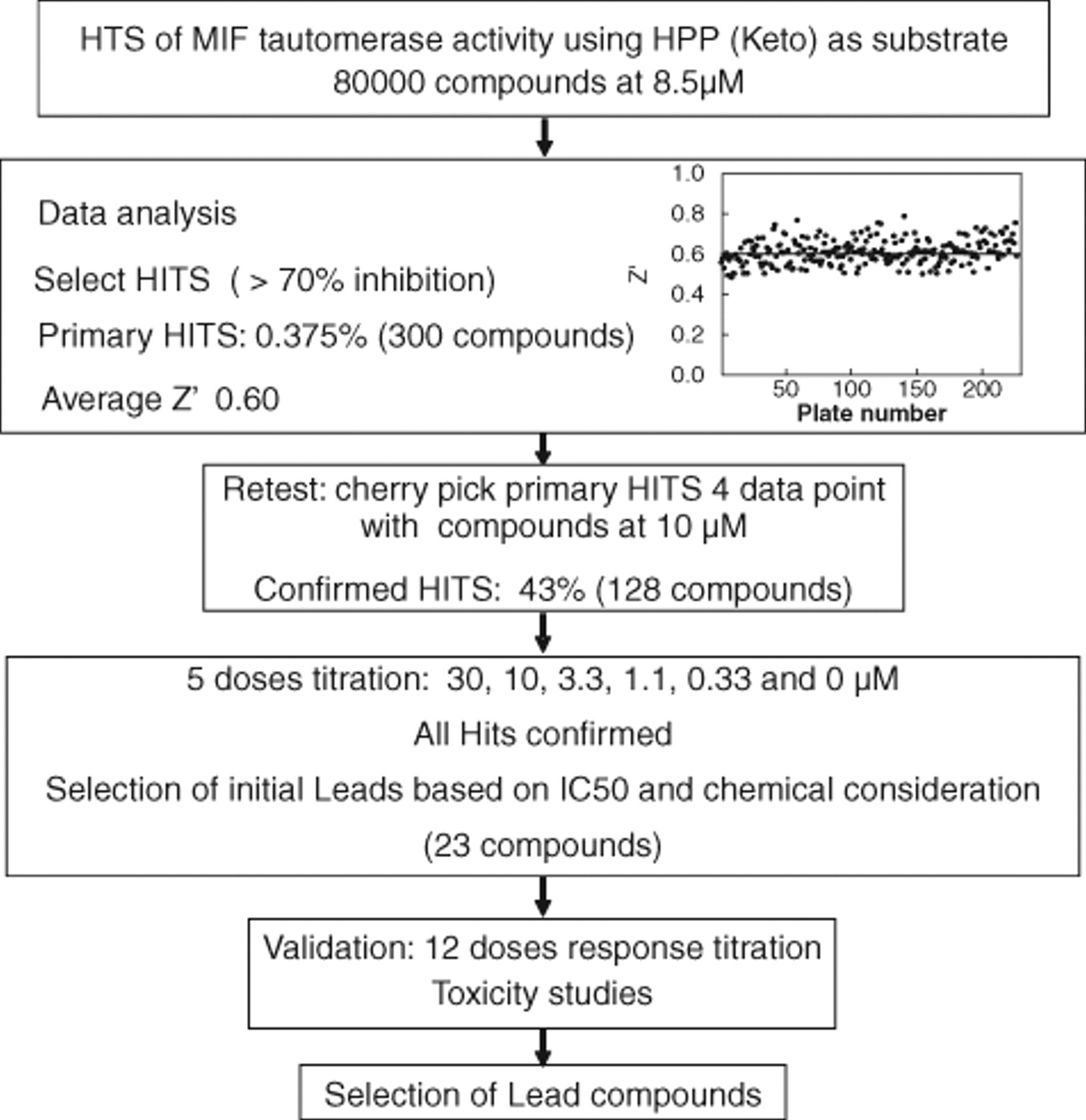
Flowchart of the screening process, including hit identification, selection, and validation. The inset shows a plot of the Z′ factor for two hundred twenty-seven 384-well plates used in the screen. The average Z′ value of the screen is 0.6 ± 0.067 (mean ± SD).
The primary hits were retested from 10-mM stock compound plates. The tautomerase assay was performed for 300 primary hits at 10 µM. Of these, 128 compounds with activities >70% were selected and retested at 6 concentrations (30, 3, 1, 0.3, 0.1, and 0 µM) in triplicate to confirm a concentration-dependent response. The most potent inhibitors were selected for further evaluation. The top 20 compounds were chosen based on activity, a “well-behaved” dose response (e.g., Hill slope = 1), and chemical class. The 20 initial lead compounds were reordered from their respective suppliers, and full dose dependence inhibition studies were performed. For each compound, 12 concentrations were carried out in triplicate, and Ki values were measured ( Fig. 6 ). Several compounds showed Ki values below 2.5 µM (MIF 1, 11, 17, and 18), and 1 lead compound, MIF 13, offered a Ki value of 500 nM. The rest of the lead compounds exhibited Ki values between 6 and 13 µM. The Ki values for hits MIF 5, 7, 8, and 15 could not be calculated as they were no longer commercially available. Therefore, close structural analogs of each compound were selected and tested. These analogs did not show any inhibition of MIF’s tautomerase activity. The table in Figure 6B shows a comparison of the Ki values from the 5-point confirmation from the screening stocks and the 12-point dose dependence inhibition studies using the resupplied compounds (reordered as powders). This comparison supports the robustness of the assay as well as the integrity of the compound stocks.
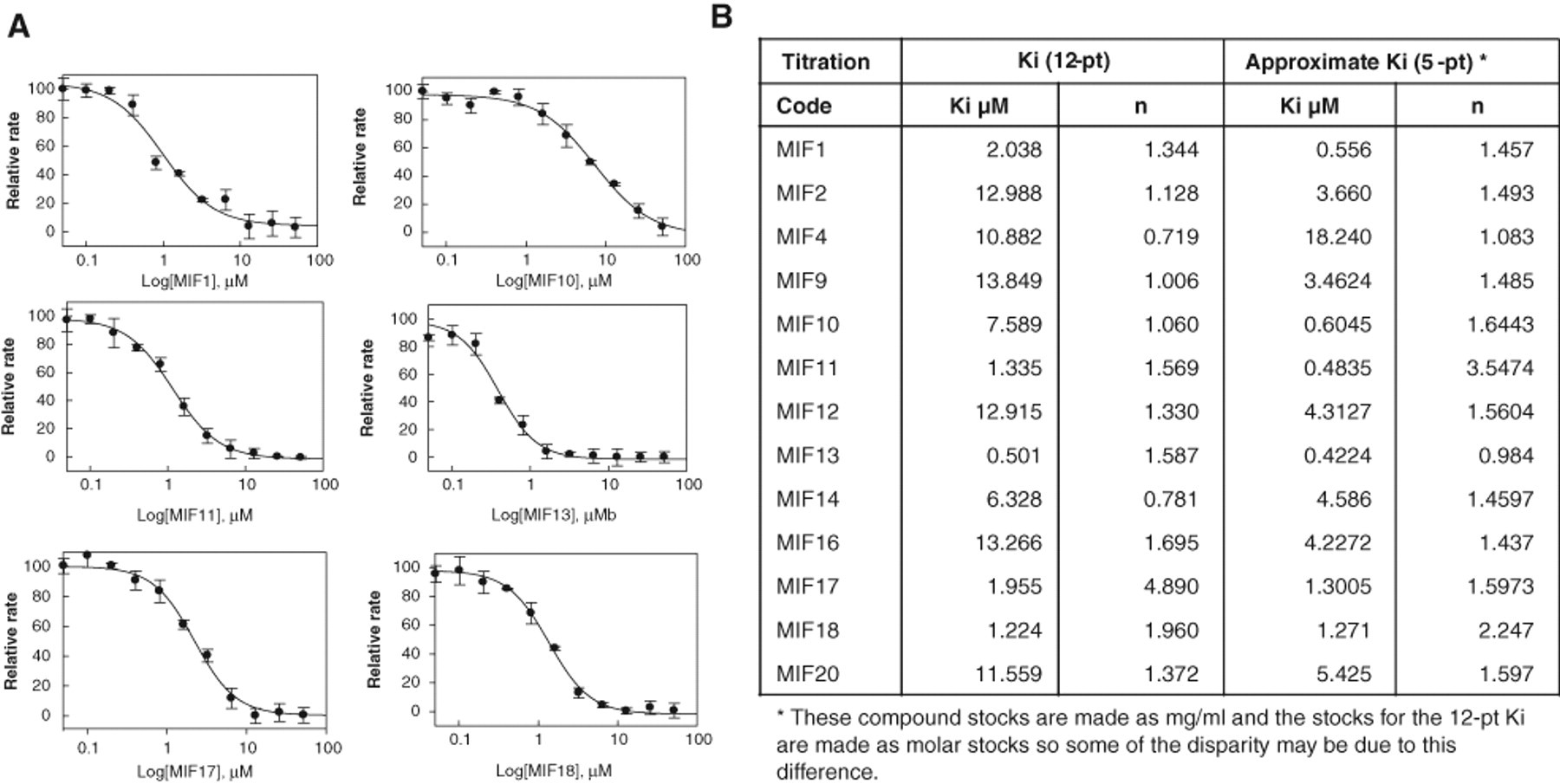
Ki value calculation of the selected lead compounds. (
HTS revealed novel classes of covalent inhibitors that act via modification of Pro-1 and Cys80
To determine the mechanism of action of the obtained hits, we first examined if tautomerase inhibition occurred via covalent modification of MIF. MIF (20 µM) in 25 mM Tris buffer (pH 7.4) was incubated with hit compounds (10 µM) for 1 h and analyzed by MALDI-TOF mass spectrometry. Under these conditions, a shift in the monomeric molecular mass of MIF by 140 mass units from 1287.79 to 1427.73 (hit compound 4) and 118 mass units from 1287.79 to 1405.76 (hit compound 12) was observed ( Fig. 7 ). At concentrations of 10 µM, we observed complete conversion to the singly modified form of MIF with hit compounds 4 and 12. To identify the site of covalent modification, the modified MIF was subjected to proteolytic digestion by trypsin and peptide mapping by mass spectrometry. Tryptic digestion of covalently modified MIF with hit compound 4 or 12 revealed a peptide fragment with an m/z ratio of 1427.73, which corresponded to the N-terminal peptide PMFIVTNTNVPR (m/z = 1287.79) plus the reactive group from hit compound 4 or 12. MS/MS analysis of this peptide demonstrated that the modification occurs at the N-terminal proline residue (Supplementary Fig. 2B,C).
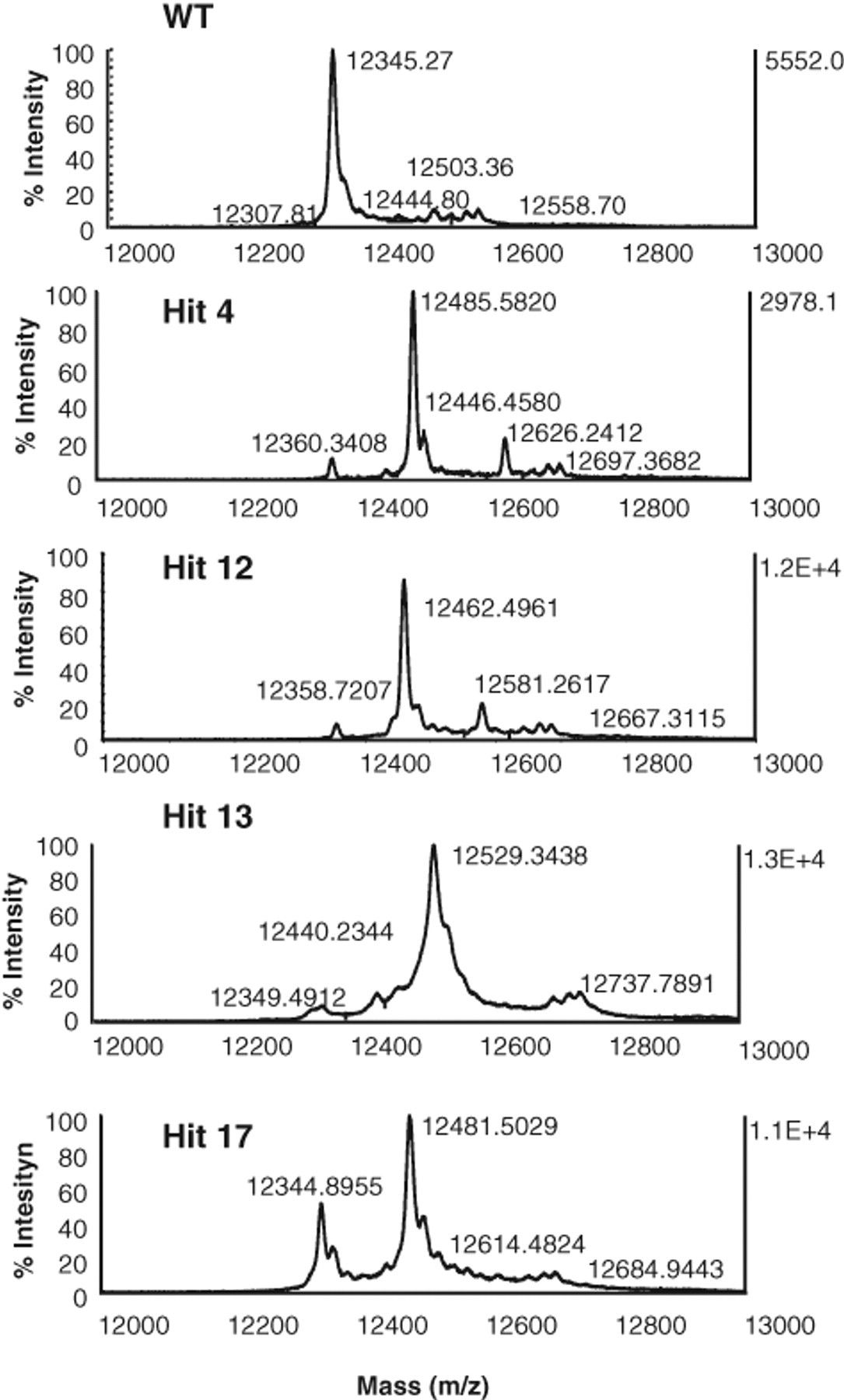
Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS) analysis demonstrates that hit compounds 4, 12, 13, and 17 covalently modify migration inhibitory factor (MIF).
Hit compounds 13 and 17 were also observed to inhibit MIF via covalent modification. A mass shift by 137 mass units from 987.55 to 1123.62 could be seen postincubation with both hit compounds 13 and 17 ( Fig. 7 ). In the case of the hit compounds 13 and 17, tryptic digestion of modified MIF revealed a new fragment peptide with an m/z ratio of 1123.62, which corresponded to the amino acid sequence spanning residues 78 to 86 (LLCGLLAER, m/z = 987.55) plus the reactive group from hit compound 13 or 17. MS/MS analysis of this peptide fragment demonstrated that the modification occurs on cysteine 80 (Supplementary Fig. 3B,C). We did not observe any covalent modifications by the remaining hit compounds, suggesting that they function via noncovalent interactions with MIF. Further studies are currently under way to elucidate the structural basis and mode of action behind their inhibitory properties.
Toxicity studies on Raw 264.7 macrophages
To determine the relative toxicity of the hit compounds, 2 assays were used to assess viability of Raw 264.7 macrophages: the MTS assay and adenosine triphosphate (ATP) luminescence-based assay. Both assays showed low toxicity for most of the hits, except MIF 16 and MIF 20, which showed toxicity at concentrations of >10 µM (Supplementary Fig. 4). Luminescence is reduced by half in the presence of 10 µM of compound 16 compared to the DMSO control, which translates to a reduction in ATP levels upon exposure to the inhibitor, a sign of unhealthy cells and compound-induced cytotoxicity.
Discussion
Inhibition of MIF activity is considered a potential therapeutic strategy for the treatment of MIF-related disorders such as sepsis, 13,14,25 rheumatoid arthritis, 26 encephalomyelitis, 27 and myocarditis. 28 In addition to circulating MIF, a significant fraction of MIF is stored in the cytoplasm and contributes to its proinflammatory activities. Therefore, strategies that target both intracellular and extracellular MIF would be more effective at inhibiting MIF and neutralizing its proinflammatory activities. Such simultaneous targeting of intracellular and extracellular MIF can only be achieved using small-molecule membrane-permeable drugs. Therefore, modulating MIF activities using small-molecule inhibitors has been of significant interest to many research groups for the past decade. In 1999, while studying the tautomerase activity of MIF, Zhang and Bucala 11 identified several D-dopachrome analogs as inhibitors of MIF tautomerase activity. Since then, many groups have focused on targeting the catalytic site, and different classes of MIF tautomerase activity inhibitors have been reported. The existing MIF inhibitors can be classified as noncovalent modifiers, such as IS01 and its analogs, 13,29,30 phenyl pyruvic derivatives, ketones, 10 and coumarin derivatives, 12 and as covalent modifiers, including NAPQI, 7 2-OBP, 31 4-IPP, 32,33 PMSF, 34 ITCs, 19,35,36 and a hydroxyquinoline. 37 These inhibitors have demonstrated complete inhibition of MIF’s tautomerase activity in vitro. In some cases, they have also resulted in the inhibition of glucocorticoids overriding activity 7 and have improved survival in an animal model of sepsis 13,14 in vivo.
The lack of a robust high-throughput activity assay has hindered the screening of chemical libraries and the discovery of diverse MIF inhibitors. Therefore, screening of large chemical libraries for MIF inhibitors has only been performed using computer-assisted searches, employing molecular dynamics, docking, and other in silico computational approaches. A virtual screen was reported by 3 different groups to discover novel inhibitors of MIF’s tautomerase activity. Orita and colleagues 10 screened 1 million compounds virtually, selected the top-ranked 524 compounds, and tested them individually in vitro. This led to the identification of 14 novels inhibitors with Ki values in the range of 0.038 and 7.4 µM. In addition, during their virtual screening, Winner et al. 32 identified 4-IPP as a covalent MIF inhibitor with an IC50 of 5 µM. Recently, Cournia et al. 38 have screened 2.1 million compounds in silico, and among 1200 top-ranked compounds, 13 were confirmed to have tautomerase inhibitory activity in the micromolar range.
The development of a high-throughput activity-based assay allows the identification of diverse classes of potent MIF inhibitors. The evidence for a direct link between MIF tautomerase activity and its physiological function remains unclear. Mutational analysis of proline1, which acts as a catalytic base, or the addition of compounds that target the catalytic site, such as IS01 or OXIME11, have been shown to attenuate MIF’s enzymatic and cytokine activities. They have also demonstrated significant protection from lethality by sepsis. 13,14 The insertion of alanine between pr01 and met2 or substituting proline with glycine not only abolishes the tautomerase activity of MIF but also blocks the induction of superoxide production in neutrophils. 8,39 On the other hand, other studies have demonstrated a preservation of proinflammatory functions for catalytically inactive MIF. 16,40 Recently, Fingerle-Rowson et al. 41 created a knock-in mouse in which the endogenous MIF gene was replaced by tautomerase null P1G. The phenotypes of wild-type (WT) and knock-in mice were compared, and an intermediate level of activity for the tautomerase null mice was observed. This demonstrates the role of protein interactions with the N-terminal region for receptor-mediated MIF signaling, in contrast to its intrinsic tautomerase activity. Therefore, targeting the tautomerase activity may not be sufficient to block MIF’s cellular functions. For this reason, we sought to develop and optimize an experimental kinetic assay for HTS that uses MIF tautomerase activity as a readout (where the inhibition could take place through different modes of action). The tautomerase catalytic site exists at the interface of 2 monomers, and the N-terminal proline plays an important role in this activity. Decreasing MIF’s enzymatic activity could occur due to (1) selective modification of the catalytic proline residue, (2) allosteric inhibition and modification, (3) dissociation of the catalytic site by trimer disruption, and (4) displacement of substrate and competitive inhibition. We hypothesized that an activity-based (tautomerase activity) HTS assay would enable the identification of all of these classes of inhibitors.
Optimizing a kinetic tautomerase assay in a high-throughput format for MIF was challenging. The tautomerase reaction of hydroxyphenyl pyruvate catalyzed by MIF is very fast: the progress curve of the reaction is linear for a maximum of 60 s. Accordingly, neither known MIF substrate possesses the required stability for an HTS assay. We optimized the assay conditions for reproducibility, with a linear range of up to 200 s. The substrate was added to the plate and mixed by orbital shaking in the reader a few seconds prior to measurement. With a goal to identify all types of inhibitors, the screening was done at a substrate concentration near its Km value to establish a balanced steady-state situation in which free enzyme and E:S complex exist in near-equal concentrations. The enzyme was preincubated with compounds for 25 min to allow the identification of slow binding and covalent inhibitors. The enzyme concentration required for robust detection of MIF activity for the HTS assay was high (400 nM), which means that potent compounds below that concentration cannot be assessed accurately. Most of the compounds identified have Ki values >1 µM. Compounds will be retested with a more sensitive plate reader at lower enzyme concentrations for more accurate Ki measurements.
In summary, using a kinetic-based assay, we were able to screen a large compound library and discover different classes of MIF inhibitors. Preliminary studies to elucidate the mode of action of these compounds revealed 3 classes of inhibitors against MIF tautomerase activity: (1) molecules that act by modifying the catalytic N-terminal pro-1 residue, (2) a novel class of molecules that act via selective modification of Cysteine 80, and (3) diverse compounds that exert their effects via noncovalent mechanisms. The mechanism of action of each compound will be confirmed using an array of biochemical and biophysical assays developed in our laboratory to probe the structure-function relationship of MIF. 19,20 Based on preliminary data, some of the inhibitors identified are reversible. These molecules will be useful as molecular tools to clearly determine whether tautomerase activity is sufficient to affect cellular function in disease models. These inhibitors may also provide leading candidates for the development of drugs to treat MIF-related diseases.
Footnotes
Acknowledgements
We thank Professor Ross Stein for his contributions to the assay development, Ms. Mickey Huang, Mr. Eli Schuman, and Mr. John Concannon from the Laboratory for Drug Discovery in Neurodegeneration for their advice and assistance, and Dr. Marc Moniatte, Mr. Diego Chiappe, and Mr. Jerome Vialaret from the proteomic core facility at the EPFL (![]() ) for their assistance with mass spectrometry data collection.
) for their assistance with mass spectrometry data collection.
This work is supported by funding to the Laboratory of Molecular Neurobiology and Neuroproteomics from the Ecole Polytechnique Federale de Lausanne (EPFL) and Swiss National Science Foundation (Grant# 310000-110027).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.