
Abstract
Assay technologies that were originally developed for high-throughput screening (HTS) have recently proven useful in drug discovery for activities located upstream (target identification and validation) and downstream (ADMET) of HTS. Here the authors investigated and characterized the biological properties of a novel target, IRE1α, a bifunctional kinase/RNase stress sensor of the endoplasmic reticulum (ER). They have developed a novel assay platform using the HTS technology AlphaScreen® to monitor the dimerization/oligomerization and phosphorylation properties of the cytosolic domain of IRE1α. They show in vitro that dimerization/oligomerization of the cytosolic domain of IRE1 correlated with the autophosphorylation ability of this domain and its endoribonuclease activity toward XBP1 mRNA. Using orthogonal in vitro and cell-based approaches, the authors show that the results obtained using AlphaScreen® were biologically relevant. Preliminary characterization of assay robustness indicates that both AlphaScreen® assays should be useful in HTS for the identification of IRE1 activity modulators.
Keywords
Introduction
L
In the present study, we used AlphaScreen® to investigate cell signaling pathways 1 and in particular those emanating from the endoplasmic reticulum (ER) under stress conditions. The ER ensures proper protein folding and export to later compartments of the secretory pathway. This is achieved through complex machineries, including protein synthesis, translocation/folding, quality control, ER-associated degradation (ERAD), and export. 2 In addition to these functional attributes, the ER has evolved a highly conserved adaptive signaling pathway, referred to as the unfolded protein response (UPR), whose activation occurs upon accumulation of improperly folded proteins in the ER. 3
UPR signaling is mediated by 3 ER resident transmembrane proteins—the PKR-like ER kinase (PERK), the activating transcription factor 6 (ATF6), and the inositol requiring enzyme 1 alpha (IRE1α). 3,4 Our work has focused on IRE1α, which is a transmembrane sensor of ER stress. The luminal domain of IRE1α contains binding sites for the chaperone BiP, whereas the cytosolic region has 2 main catalytic elements: a serine/threonine kinase and an endoribonuclease domain. 4-6 Under basal conditions, IRE1α is thought to exist as a monomer, and in response to the accumulation of misfolded proteins in the ER, IRE1 proteins oligomerize, resulting in its trans-autophosphorylation and triggering of its endoribonuclease activity. 4 However, structural studies in Saccharomyces cerevisiae showed that the luminal domain of IRE1 exists as a dimer/oligomer and suggested that it could potentially bind directly to unfolded peptides similar to MHC class I molecules. 7 This was confirmed by structural studies on the cytosolic domain of IRE1 in S. cerevisiae, which revealed a dimer conformation that is subjected to conformational changes upon ER stress, leading to activation of its kinase and RNAse activities. 8 Recently, the cytosolic domain of IRE1 was crystallized in an oligomeric form, which was promoted by the presence of the kinase inhibitor Sunitinib. 9,10 This highly ordered molecular structure was found to be physiologically relevant in S. cerevisiae. 11 Therefore, a revised model for IRE1 activation process has been proposed in which different IRE1 dimers promote the trans-autophosphorylation that is observed in vivo in oligomeric structures. This would rearrange the RNAse domain and create a binding surface for the mRNA substrates. 9 However, this model does not take into account the capacity of IRE1 to induce JNK 12 and ERK signaling cascades 13 and does not provide any insights into the mechanisms by which these kinases are activated downstream of IRE1α. 6 Regardless, IRE1α endoribonuclease activity then initiates an unconventional splicing of XBP1 mRNA that introduces a translational frame shift leading to the synthesis of a potent transcription factor. 14 Recently, IRE1 endoribonuclease activity has been shown to be partly independent of IRE1 phosphorylation, 15 suggesting the existence of alternative activation modes. 6
Thus far, the only in vitro or in vivo assays that have been used for identifying IRE1 modulators and reporting on IRE1 biological functions have mainly focused on the endoribonuclease activity of IRE1 toward XBP1 or its functional yeast homolog HAC1. 16-19 In the present study, we have used AlphaScreen® to investigate and characterize IRE1α signaling properties. We have developed 2 AlphaScreen®-based assays for IRE1α oligomerization and phosphorylation and validated the results using orthogonal in vitro and cell-based experimental systems.
Materials and Methods
Cloning
IRE1cyto cDNA (AA 470 to 977) was cloned from human liver cDNAs using either the Gateway® technology (Invitrogen, Carlsbad, CA) in pGEX-2TK or pDEST17. IRE1cyto cDNA devoid of ATG was amplified by PCR using the Platinium® Taq DNA Polymerase High Fidelity (Invitrogen) and the following amplification scheme: denaturation 94 °C for at 40 s, annealing at 60 °C for 40 s, and elongation at 68 °C for 2 min, 35 cycles. The PCR products were precipitated using PEG8000 and recombined into pDONR201 using the Gateway® BP clonase (Invitrogen). The plasmids were then transformed into competent DH5α cells and positive clones selected and sequenced. The clones were then recombined into destination vectors using LR clonase (Invitrogen).
Recombinant protein expression, purification, and quality control
The above-described plasmids were transformed into competent DH5α cells. Five individual colonies were selected and pooled, and plasmid DNA was amplified and subsequently transformed into competent BL21 bacterial cells. Recombinant protein expression in BL21 cells was induced using 1 mM isopropyl β-
Thrombin cleavage
Glutathione-S-transferase (GST)–IRE1cyto coupled to glutathione (GSH)–sepharose beads was incubated in the presence of thrombin (50 UI) for 16 h at room temperature. The supernatant was collected following centrifugation at room temperature for 10 min in a bench-top microfuge at maximal speed. Thrombin was removed following incubation with benzamidine-sepharose beads for 45 min at 4 °C. The resulting cleaved IRE1cyto was then concentrated and dialyzed using Amicon ultra centrifugal filters (cut-off = 10,000 Da; Millipore). The purified products were then suspended in 6× Laemmli sample buffer, 21 resolved by SDS-PAGE, and either directly stained using Coomassie Brilliant Blue R250 (CBB) or immunoblotted using anti-GST (Hyperomics Farma, Pierrefonds, Quebec, Canada), anti-IRE1α (Santa Cruz Biotechnology, Santa Cruz, CA), or anti-phospho-IRE1 (Abcam, Inc., Cambridge, MA) antibodies. S. cerevisiae IRE1cyto was cloned from genomic DNA and subcloned in pET15b by restriction cloning. The corresponding recombinant protein was expressed in BL21 cells and purified on nickel-coated beads NiNTA (QIAGEN, Inc., Valencia, CA) according to the manufacturer’s recommendations. Where designated, the 6xHis tag was removed using TEV protease cleavage and dialyzed/concentrated using Centricon concentrators (Millipore).
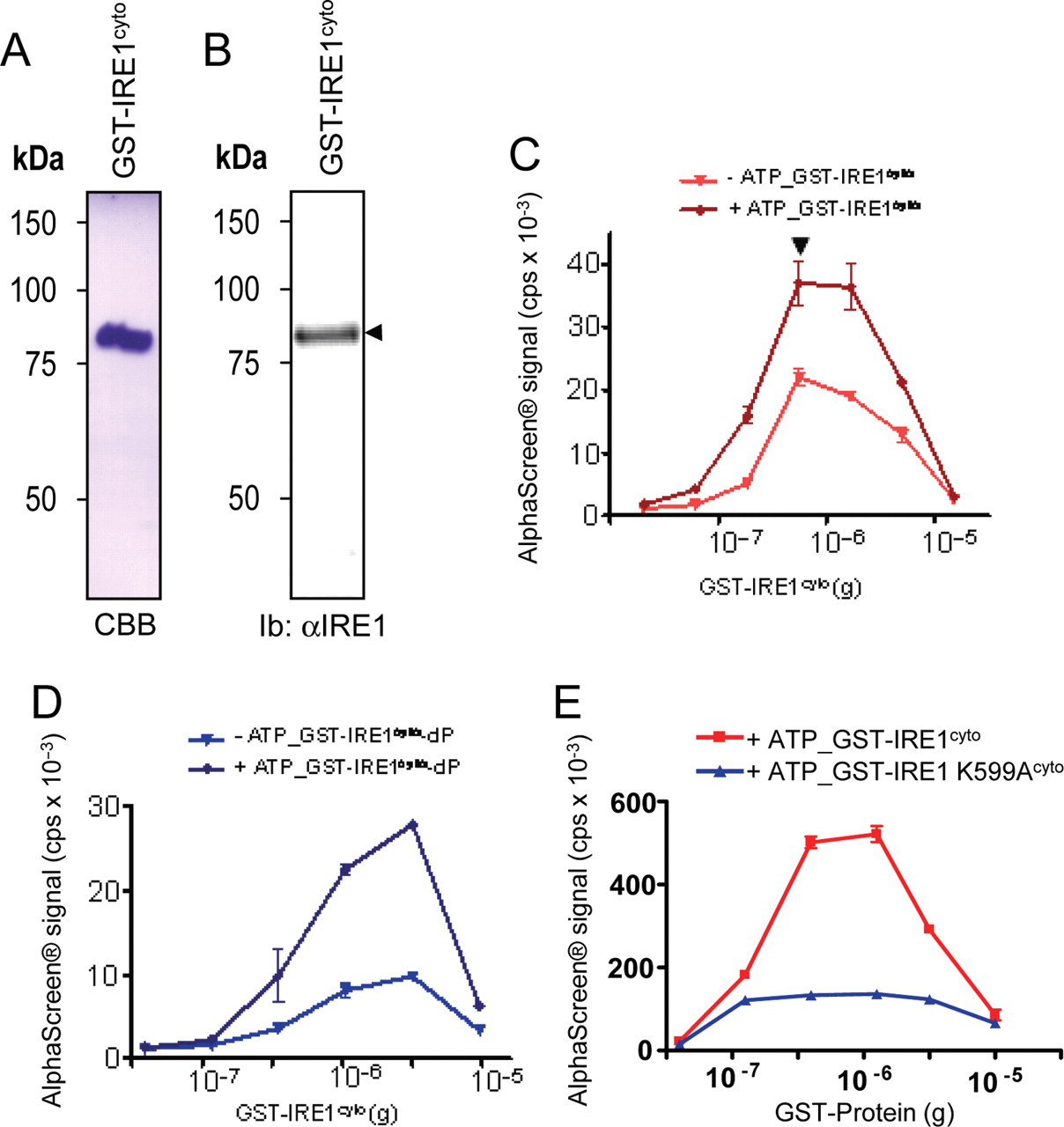
IRE1cyto AlphaScreen®-based dimerization/oligomerization assay. (
AlphaScreen®-Based Assays
AlphaScreen® assays were performed in Costar 384-well microplates in a 25-µL final reaction volume. GSH donor beads or anti-GST acceptor beads (PerkinElmer, Inc., Waltham, MA) were used at a final concentration of 0.02 mg/mL per well. The assays were performed in 20 mM Tris-HCl (pH 7.4), 50 mM NaCl, 1 mM MgCl2, 1 mM MnCl2, 2 mM dithiothreitol (DTT), and 0.1% Tween-20. All incubations were performed at 23 °C. Laser excitations were carried out at 680 nm, and readings were performed at 520 to 620 nm using the EnVision® (PerkinElmer) plate reader.
Dimer/oligomer formation assay
Increasing concentrations of GST-IRE1cyto, dephosphorylated GST-IRE1cyto, or GST were incubated for 45 min with 1 mM adenosine triphosphate (ATP; Sigma-Aldrich, St. Louis, MO) or 1 mM ATPγS (Sigma-Aldrich) or buffer and donor beads. Acceptor beads were then added to the reaction and incubated for an additional hour before reading.
Nucleotide specificity assay
Optimal concentrations of GST-IRE1cyto or dephosphorylated GST-IRE1cyto (determined in preliminary experiments) were incubated for 45 min with varying concentrations of ATP, adenosine diphosphate (ADP), adenosine monophosphate (AMP), guanosine triphosphate (GTP), guanosine diphosphate (GDP; 0-10 mM; Sigma-Aldrich), and donor beads. Acceptor beads were then added to the reaction and incubated for an additional hour before reading.
Oligomer formation assay
Optimal concentrations of GST-IRE1cyto or dephosphorylated GST-IRE1cyto were incubated for 45 min with increasing concentrations of 6xHis-hIRE1cyto, bovine serum albumin (BSA) or 6xHis-ScIRE1cyto in the presence of 1 mM ATP and donor beads. Acceptor beads were then added to the reaction and incubated for an additional hour before reading.
Phosphorylation assay
Increasing concentrations of GST-IRE1cyto were incubated for 45 min in the presence or absence of ATP. GSH donor beads and Lewis Metal–Fe acceptor beads (PerkinElmer) were added and incubated for an additional hour prior to reading. The assays were performed using thioxene, anthracene, and rubrene containing acceptor beads in all the experiments except for the experiment carried out in
RNA cleavage assay
Total RNA (10 µg) from HepG2 cells was incubated with GST-IRE1cyto (10 µg) at 37 °C for 0 to 10 h in 50 mM Tris-HCl (pH 7.5), 120 mM NaCl, 1 mM MgCl2, 1 mM MnCl2, and 5 mM β-mercaptoethanol, supplemented with 1 mM ATP or 1 mM ATPγS. Cleaved or uncleaved RNAs were used as a template for reverse transcription using XBP1 primers and GAPDH as an internal control.
In vitro phosphorylation of GST-IRE1cyto
Two micrograms of GST-IRE1cyto purified as described above were incubated for 30 min at 37 °C with 20 mM Tris-HCl (pH 7.5) in the presence of 5 to 20 mM MgCl2, 0 to 5 mM MnCl2, 2 µCi of [32Pγ]-ATP, and 100 µM ATP. The reaction was then stopped by the addition of 5 mM ATP and the reaction products analyzed by SDS-PAGE followed by Coomassie Brilliant Blue R-250 staining and radioautography. Phosphorylation was also detected using anti-phospho-IRE1 (Abcam) antibodies.
Cell culture and transfection
HeLa or Chinese hamster ovary (CHO) cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal calf serum and antibiotic. HeLa and CHO cells were transiently transfected with the pED-IRE1 wild-type (WT) plasmid or pED-IRE1 K599A plasmid. 22 Transfections were performed using LipofectAMINE (Invitrogen) according to the manufacturer’s recommendations.
In-cell cross-linking
HeLa cells transfected with pCDNA3 containing wild-type human IRE1 cDNA were washed twice with phosphate-buffered saline (PBS). The cross-linker DSS solution (final concentration 1 mM) was added and cells incubated for 2 h on ice. The reaction was stopped by the addition of 20 mM Tris-HCl (pH 7.4). Cells were lyzed with 1% Triton X-100 in 20 mM Tris-HCl (pH 7.4) buffer in the presence of protease inhibitors. Proteins were resolved by SDS-PAGE followed by immunoblot analysis using anti-IRE1α antibodies (Santa Cruz Biotechnology).
IRE1cyto in vitro cross-linking
First, 4 µM of purified IRE1cyto or dephosphorylated IRE1cyto was incubated in a final volume of 50 µL for 30 min at 4 °C. Then, the cross-linker DSS (Thermo Fisher Scientific, Inc., Waltham, MA), 1 mM final concentration, or DMSO as a control was added and the samples incubated for 2 h at 4 °C. The reaction was stopped with 20 mM Tris-HCl (pH 7.4). The samples were then suspended in 6× Laemmli buffer and the proteins resolved by SDS-PAGE. The gel was then either silver stained or subjected to immunoblot analysis using anti-IRE1α (Santa Cruz Biotechnology) or anti-phospho-IRE1 (Abcam) antibodies.
Results
Because IRE1 could be a relevant therapeutic target in many diseases, it was important to design biological activity assays that could be adaptable to high-throughput screening (HTS). To this end, we designed an in vitro assay for IRE1 using the AlphaScreen® technology to monitor its dimerization/ oligomerization and phosphorylation properties. AlphaScreen® is a technology in which donor (photosensitizer) and acceptor (chemiluminescer) microbeads can be coated with target-specific antibodies, secondary antibodies, proteins, or any molecular entity of interest. A signal is produced when the acceptor and donor beads are brought into proximity (<200 nm) by a molecular interaction occurring between the binding partners captured on the beads. Laser excitation of the Donor beads at 680 nm causes ambient oxygen to be converted to the singlet state by photosensitizers (phtalocyanine). These react with chemiluminescent agents (thioxene, anthracene, rubrene) on the acceptor bead only when the latter is in close proximity. Upon energy transfer between those compounds, activated rubrene emits light at 520 to 620 nm, which is in turn detected by the photo-detector in a microplate reader. An excitation wavelength higher than the emission wavelength ensures a low fluorescent background by avoiding any autofluorescence from biological media or compounds.
Purification of recombinant IRE1α cytosolic domain and AlphaScreen® dimerization/oligomerization assay development
We used the AlphaScreen® technology to evaluate the dimerization/oligomerization steps of human IRE1α cytosolic domains, a prerequisite for IRE1 activation.
7,8
IRE1cyto was then assayed for its ability to dimerize/oligomerize using AlphaScreen® as follows: donor beads coated with glutathione (GSH) were incubated in the presence of increasing amounts of GST-IRE1cyto, and acceptor beads coated with GSH were then added in a second incubation step in the presence or absence of 1 mM ATP. A maximal interaction signal was observed when the assay was carried out in the presence of ATP at GST-IRE1cyto concentrations of 4.1 nM (optimal concentration of GST-IRE1cyto, arrowheads,
AlphaScreen® dimerization/oligomerization assay characterization
To further characterize the assay, dimerization/oligomerization of the dephosphorylated GST-IRE1cyto was measured 17 h after the addition of excess ATP. This treatment yielded identical signal intensities when either dephosphorylated or phosphorylated GST-IRE1cyto proteins were used (
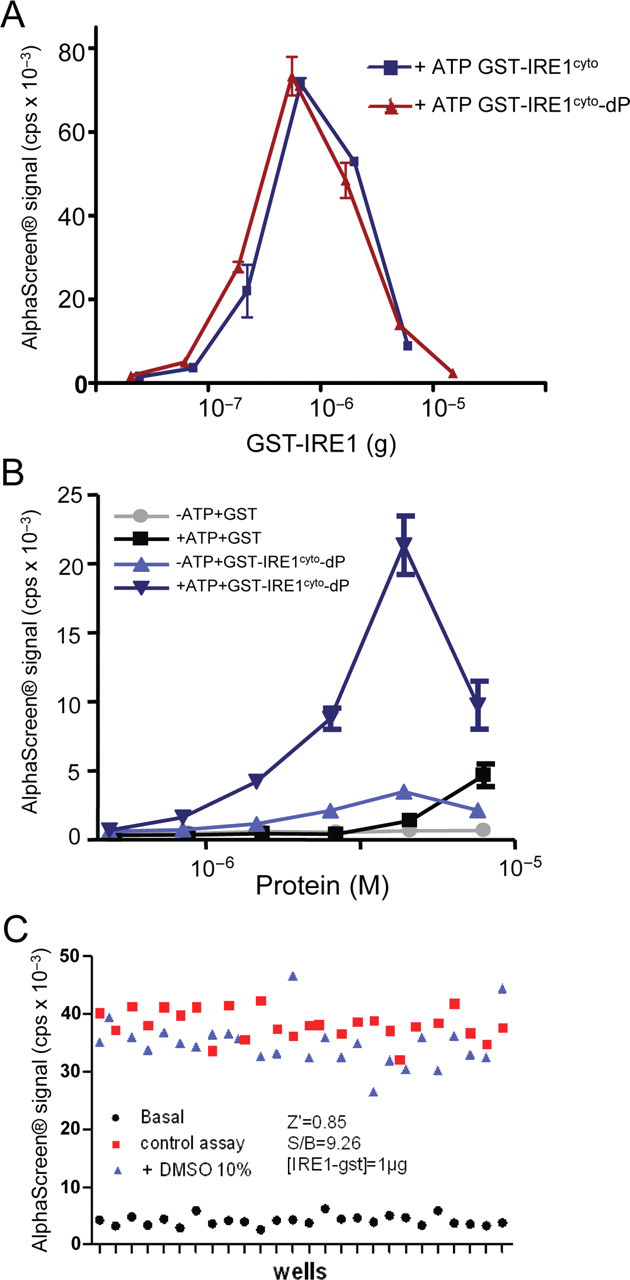
Validation and characterization of the IRE1cyto AlphaScreen®-based dimerization/oligomerization assay. (
These experiments defined a GST-IRE1cyto dimerization/oligomerization assay. Because we observed that IRE1 dimerization/oligomerization was in part dependent on its phosphorylation state, we next sought to develop an AlphaScreen®-based assay to monitor GST-IRE1cyto phosphorylation.
IRE1cyto phosphorylation properties in vitro
As our objective was to design an assay (or a combination of assays) to identify functional modulators of IRE1 activity, we first verified the biological activity of GST-IRE1cyto and tested its autophosphorylation ability in vitro as a readout using [32Pγ]-ATP (
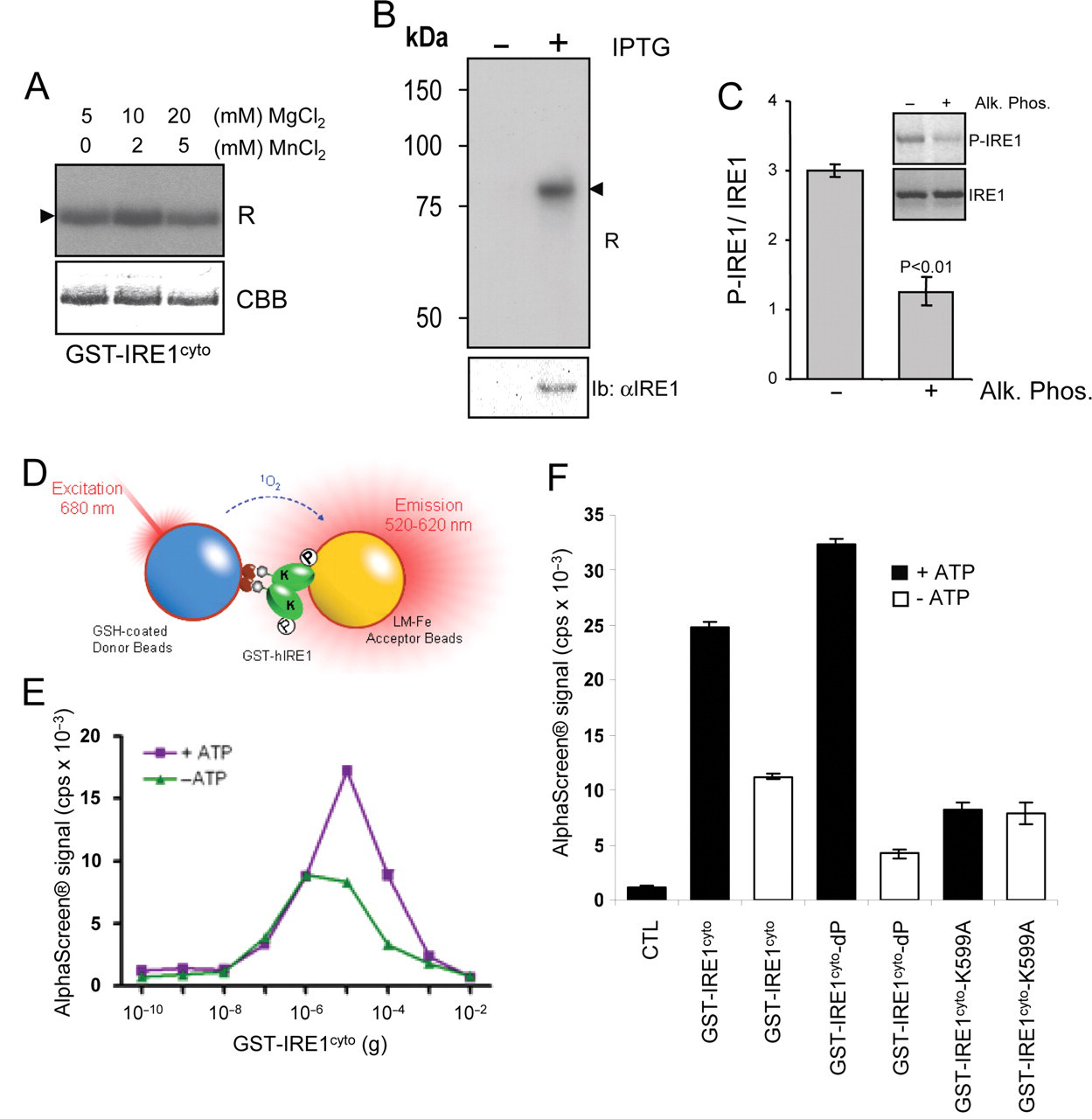
In vitro phosphorylation of IRE1cyto. (
IRE1 cytosolic domain characteristics in vitro
IRE1cyto dimer/oligomer formation was enhanced in the presence of ATP (
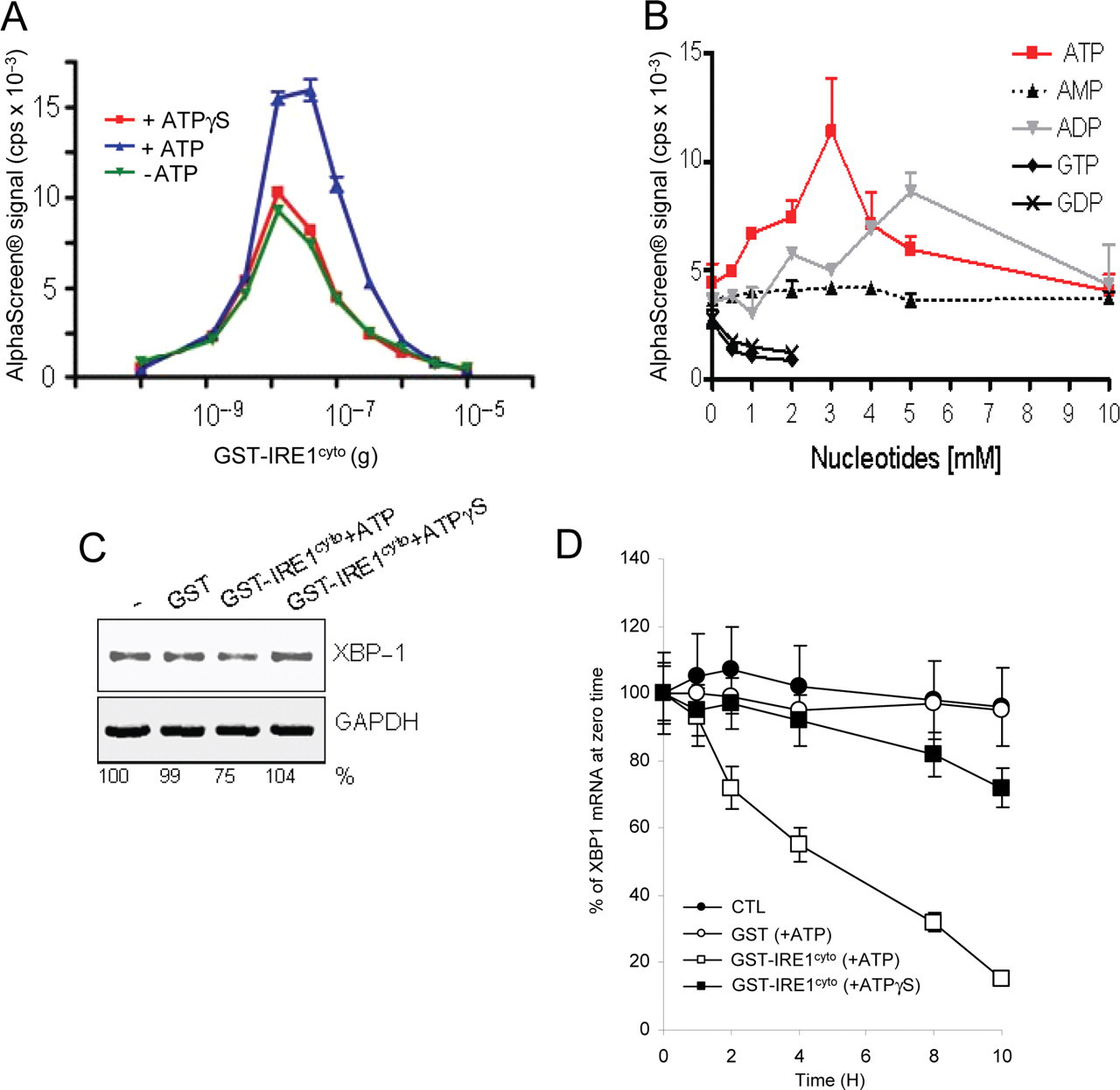
Nucleotide dependence of IRE1cyto dimerization/oligomerization and RNAse activity. (
Because IRE1 possesses a dual enzymatic activity, we next tested whether the phosphorylation and dimerization/ oligomerization status of IRE1 affected its endoribonuclease activity for XBP1 mRNA. To this end, an endoribonuclease assay was developed (see Materials and Methods), and the amount of XBP1 mRNA remaining after a 2-h incubation was monitored by RT-PCR (
Together, these experiments revealed that IRE1 dimerization/oligomerization in vitro was under the control of (1) IRE1 kinase activity, (2) nucleotide concentration, and (3) IRE1 phosphorylation status. However, these results did not completely explain the AlphaScreen® signals observed in the dimerization/oligomerization assay.
Oligomerization of IRE1 in vitro and in vivo
The above results led us to hypothesize that in our assay, the decrease in signal observed at higher GST-IRE1cyto concentrations may reflect either the loss of detectable dimers due to an excess of free GST-IRE1cyto (i.e., not bound to beads) or increased oligomer size leading to enhanced distance between donor and acceptor beads. Therefore, we further examined the ability of IRE1cyto to dimerize/oligomerize using our assay. Here, we used 6xHis-tagged IRE1cyto (
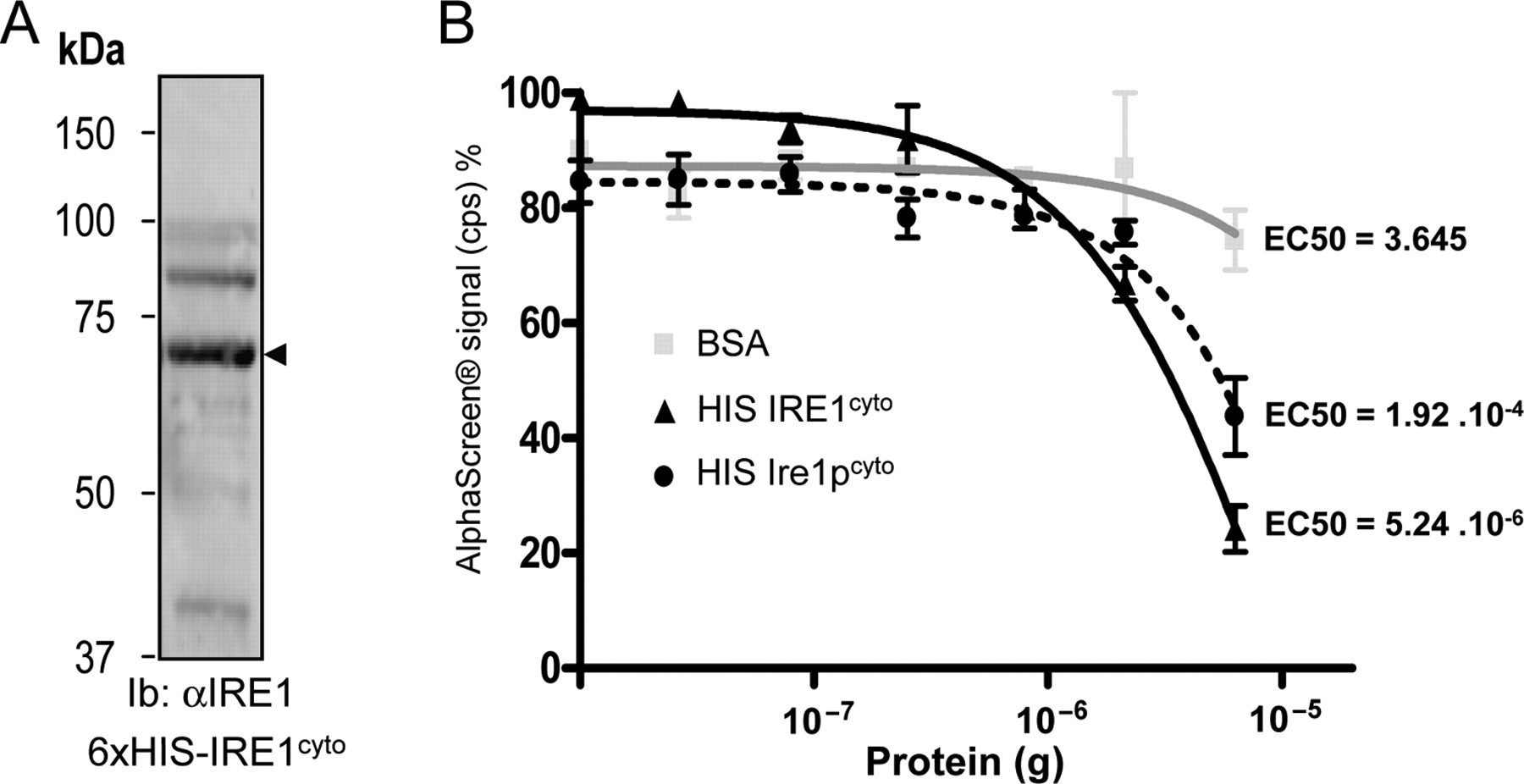
Characteristics of IRE1cyto dimer/oligomer using AlphaScreen®. (
To further analyze the mode of interaction for the IRE1 cytosolic domains, we used IRE1cyto devoid of GST (
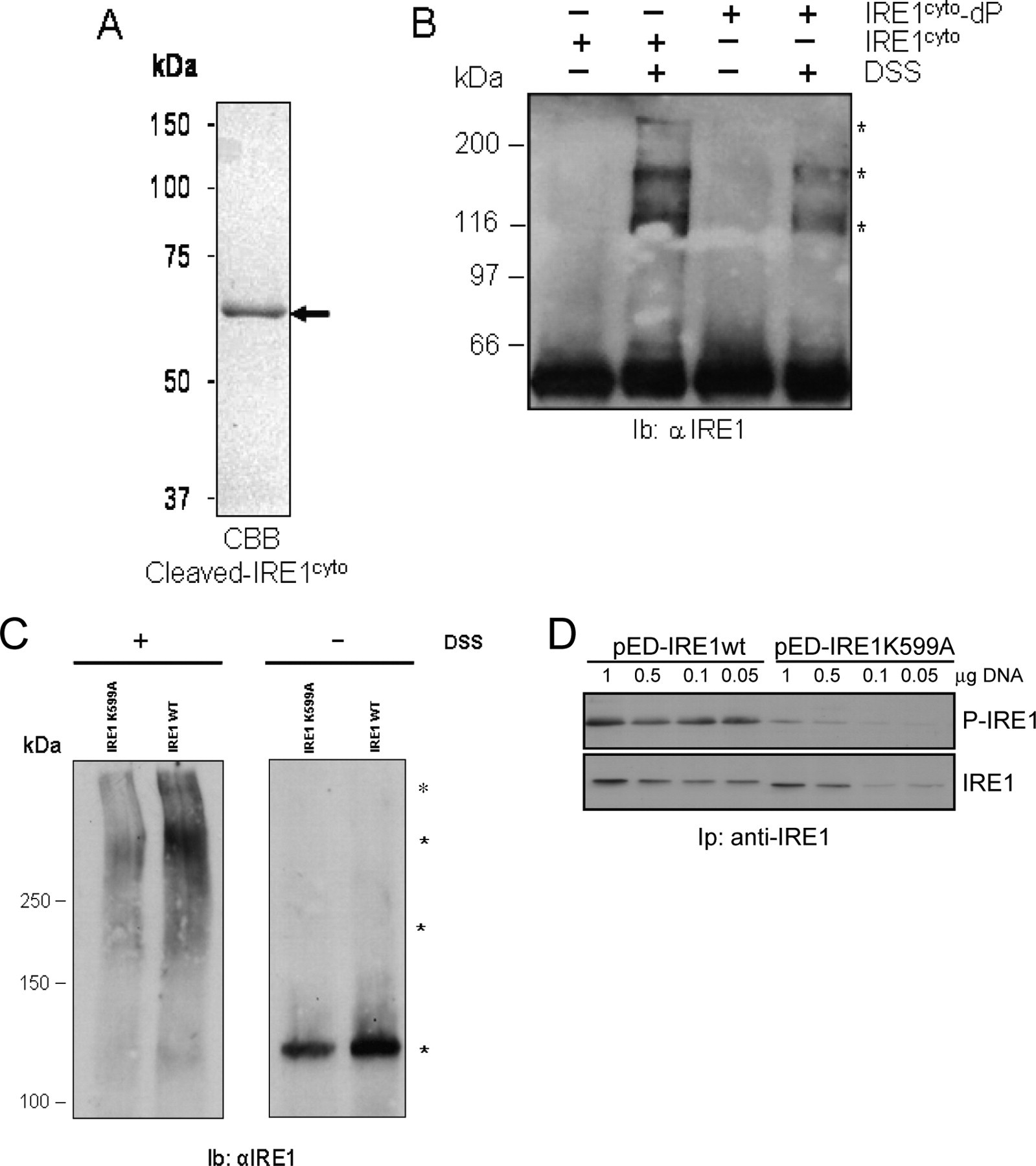
In vitro and cell-based oligomerization of IRE1. (
To test the relevance of IRE1 oligomer formation in a more physiological context, HeLa cells were transfected with a plasmid encoding wild-type human IRE1 or K599A IRE1. Following transfection, cells were subjected to chemical cross-linking using DSS. Cells were then lyzed in RIPA buffer and the resulting material resolved by SDS-PAGE followed by immunoblotting using anti-IRE1 antibodies. In the absence of DSS, only 1 immunoreactive band migrating at 110 kDa was observed (
In this work, we establish and make use of a novel AlphaScreen®-based assay to monitor IRE1 dimerization/ oligomerization properties. This was confirmed using orthogonal assays both in vitro and cell based. In addition, we developed an AlphaScreen®-based IRE1 phosphorylation assay, which could serve as a secondary screening tool to identify modulators of IRE1 activity.
Discussion
In this work, we used AlphaScreen® to investigate the biochemical properties of the dual kinase/RNAse protein IRE1. We were able to monitor and correlate 2 biochemical properties of the cytosolic domain of IRE1 (oligomerization and autophosphorylation). Moreover, we show that enhanced phosphorylation and oligomerization observed using AlphaScreen® are consistent with increased endoribonuclease activity toward XBP1 mRNA in vitro and are also relevant to events occurring in cultured cells. This provides evidence for the compatibility of using HTS technologies to address biological questions and opens new avenues to identify modulators of IRE1 activity.
Assay platform development
Thus far, the AlphaScreen® technology has been mostly used in HTS contexts in industrial settings. 1 In the past, we have used AlphaScreen® to monitor in vitro the activity of Ras GTPase, 25 but no functional significance was provided for these assays. In the present study, we focused on IRE1-mediated ER stress signaling, which is implicated in many diseases. 6 In addition, no IRE1 activity modulator has yet been reported in the literature following activity-based compound screening.
Here, our objectives were (1) to develop a series of in vitro IRE1 activity assays using an HTS technology such as AlphaScreen®, (2) to provide evidence that these assays provide biochemically relevant information on the IRE1 activation process, and (3) to demonstrate that the assays were physiologically relevant by using cell culture models. We have shown that the cytosolic domain of human IRE1α can be expressed in E. coli as 6xHis or GST fusion proteins and purified in quantities of approximately 3 mg per liter of cell culture medium. Two AlphaScreen®-based activity assays suitable for screening of IRE1 modulators were optimized. These assays were designed to monitor either the oligomerization or phosphorylation status of the cytosolic domain of IRE1. Both assays provided relative affinity values for the monomers and the autophosphorylation ability of the protein, respectively. In addition, using either the nonhydrolyzable forms of ATP, ATPγS or the catalytically inactive mutant of IRE1, IRE1 K599A, we provide evidence that both oligomerization and phosphorylation of the cytosolic domain of IRE1 are linked. We propose a model in which IRE1cyto phosphorylation may stabilize IRE1cyto oligomers.
Biological relevance of the assay platform
To confirm the information gathered from the AlphaScreen®-based experiments, we first used orthogonal in vitro assays monitoring IRE1cyto endoribonuclease activity toward XBP1 mRNA and IRE1cyto oligomerization. These assays demonstrated that the ability of IRE1cyto to form higher order oligomers correlated with its enhanced endoribonuclease activity and appeared to be dependent on IRE1cyto autophosphorylation (
In addition, these data indicate that the results obtained using AlphaScreen® were consistent with data obtained in vitro and using cell-based approaches. Moreover, because the AlphaScreen® assays provide information on IRE1cyto oligomeric and phosphorylation status and because these assays can be easily adapted for HTS, we believe that screening for IRE1 activity modulators can now be achieved. The identification of IRE1 activity modulators may certainly play significant roles in the regulation of disease progression. Indeed, it has been shown that IRE1 is involved in specific diseases involving protein misfolding and that prolonged IRE1 activation could modulate cell fate. 5,10 In addition, as IRE1 activity has already been directly implicated in tumor growth and angiogenesis, 26 inhibition of its activity may represent an interesting antitumor avenue. Finally, as illustrated by Hetz and Glimcher, 6 beyond modulation of IRE1 activity, specific targeting of IRE1-dependent signaling pathways could also constitute approaches of significant interest.
Applications to HTS
The AlphaScreen® assays are performed in a 384 format, so they are fully amenable for HTS. In addition, we have demonstrated that the oligomerization assay is not sensitive to high concentrations of DMSO (
In conclusion, we have demonstrated that the HTS technology AlphaScreen® can be used to address biological questions in an academic research setting. This may consequently represent tools not only for screening purposes but also for more fundamental investigations, which could then be easily translatable to chemical genomics-based assays.
Footnotes
Acknowledgements
We are grateful to Dr. Jean Rosenbaum, Dr. Violaine Moreau, and Ms. Pamela Cameron for critical reading of the manuscript. This work was funded by a Marie Curie International Reintegration grant; grants from the Fondation pour la Recherche Médicale, la Ligue contre le Cancer; an Avenir Program grant (Inserm); and a grant from the Institut National du Cancer to EC. MB was supported by a Ph.D. fellowship from the Conseil Régional d’Aquitaine and a fellowship from the Fondation pour la Recherche Médicale.