
Abstract
Endoplasmic reticulum (ER) stress leads to activation of caspase-12, which in turn can lead to activation of caspase-3 and cell death. Here we report that transient acidosis induces ER stress and caspase-12-mediated cell death in mouse astrocytes. After a 3-hour incubation at pH 6.0, astrocytes exhibited delayed cell death associated with nuclear condensation and fragmentation. Cell death was reduced by the protein synthesis inhibitor cycloheximide, further suggesting an active cell death program. Acidosis increased the expression of the ER chaperone protein GRP-78, indicative of ER stress. Acidosis also increased caspase-12 mRNA expression, caspase-12 protein expression, cleavage of caspase-12 to its active form, and activation of caspase-3. Each of these effects was suppressed in astrocytes pretreated with caspase-12 antisense phosphorodiamidate morpholino oligodeoxynucleotides (PMOs). Caspase-12 antisense PMOs also reduced the cell death induced by acidosis. Immunoprecipitation studies showed dissociation of both caspase-12 and Ire1-α from GRP-78, thereby suggesting a mechanism by which acidosis can initiate the ER stress response. To evaluate caspase-12 activation in vivo, rats were subjected to middle cerebral artery ischemia–reperfusion. Immunostaining of brain sections harvested 24 hours later showed increased caspase-12 expression and nuclear condensation in astrocytes of the periinfarct region exposed to acidosis during ischemia. These findings suggest that acidosis induces ER stress and caspase-12 activation, and that these changes may contribute to delayed cell death after ischemia.
Introduction
Endoplasmic reticulum (ER) stress results from a mismatch between ER protein load and ER protein-folding capacity (Harding and Ron, 2002). The ER stress response can promote either cell survival or cell death, depending on the degree of injury and other factors (Harding and Ron, 2002; Paschen, 2003; Rao et al, 2004). Acidosis is a major factor contributing to cell death after cerebral ischemia (Rehncrona et al, 1981; Kraig et al, 1986, 1987; Siesjo, 1988; Nedergaard et al, 1991; Katsura et al, 1994). Cerebral artery occlusion produces acidosis in regions of incomplete ischemia where residual blood flow carries glucose but not oxygen, leading to anaerobic glycolysis and the production of lactic acid (Nedergaard et al, 1986; Peek et al, 1989; Obrenovitch, 1995; Swanson et al, 1997). Extracellular pH in these regions can decrease to as low as pH 6.8 to 5.9, depending on the plasma glucose concentration (Kraig et al, 1986; Smith et al, 1986). Astrocyte intracellular pH may decrease to even lower (Kraig et al, 1986; Kraig and Chesler, 1990). Acidosis of this degree causes protein misfolding in astrocytes and other cell types, as evidenced by the induction of heat shock proteins (Massa et al, 1996; Narasimhan et al, 1996). Acidosis in this range also can reduce the activity of ER Ca2+ ATPase (Wolosker et al, 1997). Since protein misfolding and reduced ER Ca2+ are both known to cause ER stress (Kozutsumi et al, 1988; Paschen and Frandsen, 2001), acidosis could be an important factor contributing to the ER stress response after ischemia.
Normally, the ER chaperone protein GRP-78 binds to Ire1-α (Bertolotti et al, 2000) and procaspase-12 (Rao et al, 2002b) on ER membranes. During ER stress, GRP-78 binds to unfolded proteins in the ER lumen, and both Ire1-α and procaspase-12 are released. The released Ire1-α interacts with transcription factors and other proteins to induce the changes in gene expression characteristic of ER stress (Bertolotti et al, 2000; DeGracia et al, 2002). The released procaspase-12 is subsequently cleaved to its active caspase-12 form by calpain, caspase-7, or by caspase-12 itself (Nakagawa and Yuan, 2000; Rao et al, 2001; Fujita et al, 2002). Caspase-12 can activate caspase-9 by a mechanism that is independent of cytochrome c (Morishima et al, 2002; Rao et al, 2002a), which in turn may induce caspase-3 activation and cell death. Caspase-12 may also promote cell death by other routes (Oubrahim et al, 2002).
Acidosis has not previously been identified as a factor inducing ER stress or caspase-12 activation. Astrocytes are the most numerous cell type in mammalian brain and perform several functions that are critical for normal brain function and neuron survival (Chen and Swanson, 2003). Astrocytes might be as vulnerable to acidosis as neurons, or more so (Plum, 1983; Goldman et al, 1989; Giffard et al, 1990a, 2000). Since astrocytes are essential for neuronal survival, acidosis-induced astrocyte death in regions of incomplete ischemia may transform selective neuronal injury into pan-necrosis (infarction) (Plum, 1983; Chen and Swanson, 2003). In this study, we used primary murine astrocytes to determine the effects of transient acidosis on the ER stress response, caspase-12 expression, caspase-12 activation, and cell survival. Results of these studies suggest that caspase-12 contributes to delayed astrocyte death after acidosis, and that acidosis activates the ER stress pathway by inducing dissociation of procaspase-12 and Ire1-α from GRP-78. In addition, immunostaining of rat brain sections after ischemia–reperfusion showed increased caspase-12 expression in both astrocytes and neurons in the peri-infarct region that is subjected to acidosis during ischemia. These findings suggest that acidosis can stimulate the ER stress response and induce caspase-12 activation, and that these changes may contribute to delayed cell death.
Experimental Procedures
Cell Cultures
Primary astrocyte cultures were prepared from cortices of 1-day-old Swiss-Webster mice (Simonsen, Gilroy, CA, USA) as described previously (Swanson et al, 1997), in accordance with a protocol approved by the San Francisco Veterans Affairs Medical Center animal studies committee. In brief, the cortices were dissociated with papain and trituration, suspended in Eagle's minimal essential medium (MEM) containing 5 mmol/L glucose and supplemented with 2 mmol/L glutamine, 100 nmol/L sodium selenate, and 200 nmol/L α-tocopherol, and 10% fetal bovine serum (FBS) (Hyclone, Ogden, UT, USA). The cells were plated in 24-well Falcon culture plates and maintained in a humidified, 5% CO2 incubator at 37°C. The astrocytes reached confluence and ceased proliferation at days 12 to 15 in vitro. The cultures were treated on day 16 with 20 μmol/L cytosine arabinoside for 48 hours to eliminate contaminating microglia and other proliferating cell types. The cultures were subsequently maintained in MEM containing 5 mmol/L glucose and 3% FBS until use on days 21 to 24. Cultures prepared in this manner show greater than 95% immunoreactivity for the astrocyte-specific marker, glial fibrillary acidic protein (GFAP).
Incubations with Endoplasmic Reticulum Stress Agents and Low pH
Experiments were initiated by replacing the culture medium with a balanced salt solution (BSS) containing (in mM): KCl, 3.1;. NaCl, 134; CaCl2, 1.2; MgSO4, 1.2; KH2PO4, 0.5; NaHCO3, 15.7; and glucose, 2. The pH of the BSS was adjusted to 7.2 while the solution was equilibrated with 5% CO2 at 37°C. Osmolarity was verified at 285 to 300 mOsm with a Wescor vapor pressure osmometer (Logan, UT, USA). After washing with BSS, the cultures intended for acidosis treatment were then incubated for 1 to 3 hours at 37°C, 5% CO2 in BSS containing 5 mmol/L piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) and equilibrated at the designated pH value. Acidosis was terminated by washing again with BSS and returning the cultures to MEM containing 3% FBS. Control wells underwent the same medium exchanges but were incubated at pH 7.2, the normal pH of brain extracellular fluid. The ER stress-inducing agents tunicamycin (Sigma, Saint Louis, MO, USA), brefeldin A (Calbiochem, San Diego, CA, USA), and thapsigargin (Sigma, Saint Louis, MO, USA) were added to astrocyte cultures in MEM/3% FBS culture medium and incubated at 37°C, 5% CO2 until analyzed.
Lactate Dehydrogenase (LDH) Assay for Cell Survival
Astrocyte survival was determined by measuring LDH activity in cultures lysed in 5 mmol/L HEPES, 1 mmol/L dithiothreitol, and 0.1% Triton X-100 (Swanson et al, 1997). Lactate dehydrogenase activity was determined by the method of Koh and Choi (1987), using a kinetic plate reader (Molecular Devices, Menlo Park, CA, USA). Percent cell death in each well was calculated as (1−(LDH activity)/(control LDH activity)) × 100, with the control LDH activity defined as the mean value obtained in four control wells from the same 24-well plate as the test well. Control wells were treated with medium exchanges only, which causes no, or negligible, astrocyte death. Prior experience shows plating density to be nearly identical among wells within a given 24-well plate.
4,6-Diamidino-2-Phenylindole (DAPI) Staining
Morphologic changes of apoptosis were analyzed by staining cultured astrocytes for 10 minutes with the fluorescent dye DAPI (Sigma, Saint Louis, MO, USA) used at 2 μg/mL. Staining was assessed using excitation at 350 nm.
RNA Isolation and Reverse Transcription-Polymerase Chain Reaction (RT-PCR) for Caspase-12 Amplification
Total RNA was isolated from astrocytes using the TRI Reagent and protocol from Molecular Research Center (Cincinnati, OH, USA). Reverse transcription-polymerase chain reaction (RT-PCR) was performed using Reverse Transcription System (Promega, Madison, WI, USA) and oligonucleotides (Biosource International, Camarillo, CA, USA) following the manufacturer's instructions. PCR reactions contained 0.05 U/μL of Taq DNA polymerase (Roche, Penzberg, Germany) and 0.4 μmol/L each of the forward and reverse caspase-12 primers, 5′-GCCAGGAGGACACATGAAAG-3′ and 5′-GCATCTGGGTCAGTTCACCT-3′, respectively. The forward and reverse glyceraldehyde-3-phosphate dehydrogenase (G3PDH) primers were as follows: 5′-TCCCATCACCATCTTCCA-3′ and 5′-CATCACGCCACAGCTTTC-3′. PCR was performed for 30, 32, 34, 36, 38, and 40 cycles of 95°C for 1 minute, 55°C for 1 minute, and 72°C for 2 minutes. The amplified RT-PCR products were separated by 1.5% agarose gel electrophoresis and visualized by ethidium bromide staining. Because both the caspase-12 and the G3PDH bands showed a near-linear increase through 32 to 40 cycles, 36 cycles was chosen for data analyses.
Western Blotting
Cells were lysed in 5 mmol/L HEPES, 1 mmol/L dithiothreitol, and 0.1% Triton X-100 with 10 μg/mL leupeptin, 10 μg/mL pepstatin A, 10 μg/mL aprotinin, and 0.2 mmol/L phenylmethanesulfonyl fluoride (PMSF). Protein content was determined using the Bradford assay method (Biorad, Philadelphia, PA, USA) (Bradford, 1976). Sample loading buffer (75 mmol/L Tris-HCl (pH 6.8), 15% glycerol, 1.5% sodium dodecyl sulfate (SDS), 0.16 mol/L sucrose, 0.5 mmol/L EGTA, 2.5 mmol/L NaN3, 7.5% β-mercaptoethanol, and 0.00375% bromophenol blue) was added to each sample at two times the volume of the sample lysate. The mixture was then boiled for 3 minutes. Equal amounts of total cellular protein were separated by 10% SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membranes were treated for 1 hour with a blocking buffer containing a 1:1000 dilution of anti-rabbit IgG antibody conjugated with horseradish peroxidase that had been inactivated with sodium azide (Richardson et al, 1983), and for 1 hour with 5% milk. Membranes were incubated overnight at 4°C with a 1:1000 dilution of rabbit polyclonal anti-caspase-12 antibody (Cell Signaling Technology, Beverly, MA, USA), a 1:200 dilution of rabbit polyclonal anti-GRP-78 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), or a 1:400 dilution of rabbit anti-Ire1-α antibody (Zymed, San Francisco, CA, USA) in 1% bovine serum albumin/0.5% Tween-20/10 mmol/L Tris-buffered saline, pH 7.4, with 0.02% sodium azide. After washing, the membranes were incubated with a 1:2000 dilution of horseradish peroxidase-conjugated anti-rabbit IgG antibody for 2 hours at room temperature. The membranes were again washed, treated with the chemiluminescent substrate Western Lightning Chemiluminescence Reagent PLUS (Perkin-Elmer, Boston, MA, USA), and immediately exposed to Kodak X-OMAT AR film. To adjust for protein loading, membranes were also immunostained with a 1:1000 dilution of mouse monoclonal anti-β-actin antibody (Sigma, Saint Louis, MO, USA) and then with a 1:1000 dilution of horseradish peroxidase-conjugated anti-mouse IgG antibody (Vector, Burlingame, CA, USA). Optical densities of the protein bands were measured using the NIH ImageJ software program. The protein band densities were normalized in each case to the density of the β-actin band from the same sample. The normalized band densities were then expressed as a percent of controls run on the same immunoblot. Each value determined in this way was considered an ‘n’ of 1.
Immunoprecipitation
Astrocyte lysates in RIPA buffer were preincubated with 10 μL of protein A agarose (Oncogene, Boston, MA, USA) for 3 hours at 4°C to remove nonspecific binding proteins. After centrifugation at 12,000g for 20 seconds, 300 μL of supernatant was incubated with 10 μL of anti-GRP-78 antibody overnight at 4°C and then with 10 μL of protein A agarose for 3 hours at 4°C. Pellets were precipitated by centrifugation at 12,000g for 20 seconds and washed 3 times with RIPA buffer. After boiling for 3 minutes to dissociate the immune complexes, the samples were again centrifuged at 12,000g for 20 seconds and 100 μL of the supernatant was used for Western blots.
Incubations with Protein Synthesis Inhibitor or Pan-Caspase Inhibitor
For studies of acidosis, astrocytes were pretreated for 30 minutes with 1 μg/mL cycloheximide (Sigma, Saint Louis, MO, USA) or for 3 hours with 50 μmol/L of the pan-caspase inhibitor carbobenzoxy Val-Ala-Asp-fluoromethylketone (zVAD-fmk, Calbiochem, San Diego, CA, USA) and then incubated at pH 6.0. for 3 hours in the continued presence of these agents. zVAD-fmk-treated astrocytes were also incubated with 50 μmol/L zVAD-fmk after acidosis. For studies using tunicamycin, astrocytes were pretreated with 50 μmol/L zVAD-fmk for 30 minutes and coincubated with 50 μmol/L zVAD-fmk and 100 μg/mL tunicamycin for the entire 80-hour incubation period.
Caspase-12 Antisense Treatment
Preliminary studies screened four siRNA sequences and two antisense phosphorodiamidate morpholino oligodeoxynucleotide (PMO) sequences (Iversen, 2001) and found the PMO sequences to be more effective than any of the siRNA sequences examined (data not shown). The PMOs were synthesized by Gene Tools (Philomath, OR, USA). The two antisense sequences used both targeted the murine caspase-12 gene immediately upstream and overlapping the 5′ end of the open reading frame (Van de Craen et al, 1997). These sequences were 5′-TGTCCTCCTGGCCGCCATGGCTGT-3′, as previously used by Nakagawa et al (2000); and the overlapping sequence 5′-CATGGCTGTGCTCGTGCCGCTCGTG-3′. The two sequences were equally effective in suppressing caspase-12 protein expression, and all results presented here were obtained using the second sequence. The control, random sequence antisense PMO was 5′-CCTCTTACCTCAGTTACAATTTATA-3 (Boguslavsky et al, 2003). For antisense delivery, the culture medium was replaced with fresh MEM containing 10% FBS for overnight incubation. In all, 2.5 μmol/L of either the caspase-12 antisense or random oligonucleotide were combined with 0.2 μmol/L ethoxylated polyethylenimine (EPEI) (Gene Tools, Philomath, OR, USA) in BSS. The resulting antisense PMO/EPEI solution was allowed to complex for 15 minutes at room temperature, added to the culture medium, and incubated for 3 hours at 37°C in a 5% CO2 incubator. The cultures were then washed with BSS and replaced in MEM/10% FBS. Exposure to acidosis or ER stress agents was initiated 6 hours after washout of the antisense PMOs.
Caspase-3 Activity Assay
Caspase-3 activity was measured as described by Sordet et al (2002), with slight modifications. In brief, astrocytes were lysed in modified RIPA buffer containing 50 mmol/L Tris-HCl, pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L PMSF, 1 mmol/L Na3VO4, 1 mmol/L NaF, and 1 μg/mL each of aprotinin, leupeptin, and pepstatin. Protein concentrations were measured using the bicinchoninic acid method (Smith et al, 1985). Samples were loaded into 96-well black-wall plates, and a reaction mixture containing 100 μmol/L fluorogenic peptide substrate Ac-Asp-Glu-Val-Asp-AMC (DEVD-AMC; Calbiochem, San Diego, CA, USA), 1 mmol/L EDTA, 0.1% CHAPS, 10% glycerol, 20 mmol/L dithiothreitol, and 100 mmol/L HEPES (pH 7.0) was added to each sample well. After incubation for 4 hours at 37°C, fluorescence was measured using a fluorescence plate reader (Molecular Devices, Sunnyvale, CA, USA) at an excitation wavelength of 355 nm and an emission wavelength of 460 nm. Activity was calculated as the increase of fluorescence relative to that of control (pH 7.2) astrocytes and normalized to protein concentrations.
Endoplasmic Reticulum Stress Protein Expression After Brain Ischemia
Male Sprague–Dawley rats, weighing approximately 300 g, were obtained from Charles River (Wilmington, MA, USA). Transient cerebral ischemia was induced by the Hsu method (Liu et al, 1989). In brief, rats were anesthetized with isoflurane/nitrous oxide, intubated, and ventilated with a small animal ventilator. The right middle cerebral artery was exposed and ligated. Both common carotid arteries were then exposed and occluded for 1 hour using microvascular clips (Fine Science Tools, Foster City, CA, USA). Wounds were closed, anesthesia discontinued, and the rats were returned to their cages. After 24 hours, the post-ischemic or sham-operated rats were anesthetized and perfused with 4% paraformaldehyde. Brains were removed, fixed in 4% paraformaldehyde for 1 hour, and immersed for 24 hours in phosphate buffered 135 mmol/L NaCl (PBS) containing 30% sucrose. Coronal 50-μm sections were prepared and placed in PBS containing 2% sheep serum/0.2% Triton X-100/0.1% bovine serum albumin for 30 minutes at room temperature. After washing with PBS, the slices were incubated overnight at 4°C with either a 1:100 dilution of rabbit polyclonal anti-caspase-12 antibody or a 1:250 dilution of rabbit polyclonal anti-GRP-78 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and with either a 1:500 dilution of mouse anti-GFAP antibody (Chemicon International, Temecula, CA, USA) or a 1:200 dilution of mouse monoclonal antimicrotubule associated protein-2 (MAP-2) antibody (Chemicon, Temecula, CA, USA). After washing with PBS, the slices were incubated for 2 hours with a 1:250 dilution of Alexa Fluor 594-conjugated goat anti-rabbit IgG for caspase-12 and GRP-78, or a 1:500 dilution of Alexa Fluor 488-conjugated goat anti-mouse IgG for GFAP and MAP-2 (Molecular Probes, Eugene, OR, USA). Some sections were counterstained by incubation for 10 minutes with 2 μg/mL DAPI as described for the cell cultures. The sections were mounted with ProLong Antifade Kit (Molecular Probes, Eugene, OR, USA) and photographed using a Leica inverted laser-scanning fluorescent microscope equipped with a Hamamatsu ORCA CCD camera using Openlab software (Improvision). Emission signal was acquired with filters for green (FITC, 488 nm) and red (Texas Red, 594 nm) simultaneously. Emission signals from sections of 10 nm optical thickness were obtained four times and averaged to obtain the images. A standard fluorescence microscope equipped with UV excitation was used to photograph the DAPI-stained sections, using 360 nm excitation and a 420 nm barrier filter. In these sections, the Alexa Fluor 488 and Alexa Fluor 594 antibodies were used to detect anti-GFAP antibody and anti-caspase-12 antibody, respectively. Negative controls were prepared by omitting the primary antibodies. Adjacent sections were stained with hematoxylin and eosin using standard procedures (Garcia et al, 1993).
Statistical Analysis
Treatment groups were compared using one-way ANOVA followed by Fisher's protected least significant difference test for multiple group comparisons. Quantitative data are presented as means±s.d.
Results
Transient exposure (1 to 3 hours) to acidosis in the range of pH 6. 4 to 6.0 led to astrocyte death (Figures 1A and 1B). Predictably, the extent of cell death correlated with the duration and severity of the acidosis exposure. Each condition that produced significant cell death at 24 hours after acidosis produced greater cell death when assessed at 48 hours after acidosis, indicative of a delayed cell death process. DAPI staining of nuclear morphology showed progressively increased chromatin condensation and formation of apoptotic bodies over this time interval (Figure 1C), consistent with an apoptotic form of cell death. Cell death was not appreciably increased at time points later than 48 hours (not shown).
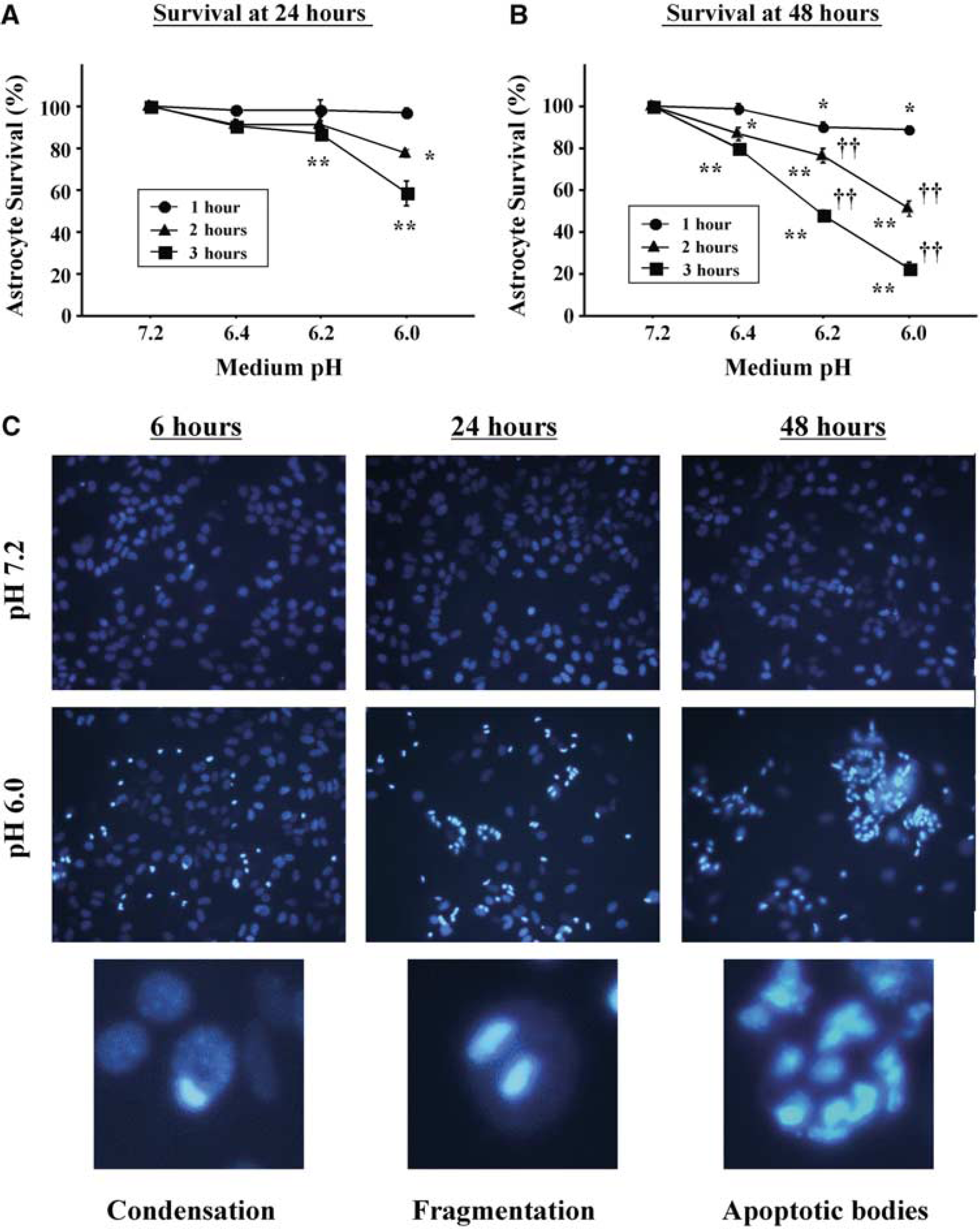
Delayed astrocyte death after acidosis. (
Acidosis Upregulates Astrocyte GRP-78 and Caspase-12 Expression, Induces Cleavage of Caspase-12 to its Active Form, and Induces Caspase-Mediated Cell Death
The effect of transient acidosis (pH 6.0) on caspase-12 mRNA expression was assessed by RT-PCR. A maximal transcript expression of roughly twice the baseline value was observed 3 hours after acidosis, with no change in the G3PDH mRNA run in parallel (Figure 2). Western blotting showed that acidosis exposure increased levels of procaspase-12 as well as the active 46 and 35 kDa caspase-12 fragments, with peak expression levels at 6 to 12 hours after acidosis (Figures 3A and 3C). Increased expression of the ER chaperone protein GRP-78 was also observed, consistent with an ER stress response (Kozutsumi et al, 1988). Astrocytes incubated with agents known to induce ER stress, tunicamycin, brefeldin A, and thapsigargin (Nakagawa et al, 2000), showed a similar pattern of increased expression of procaspase-12 and the active caspase-12 fragments (Figures 3B and 3D). Taken together, these results suggest that increased caspase-12 expression and activation are both induced by acidosis as part of the astrocyte ER stress response.
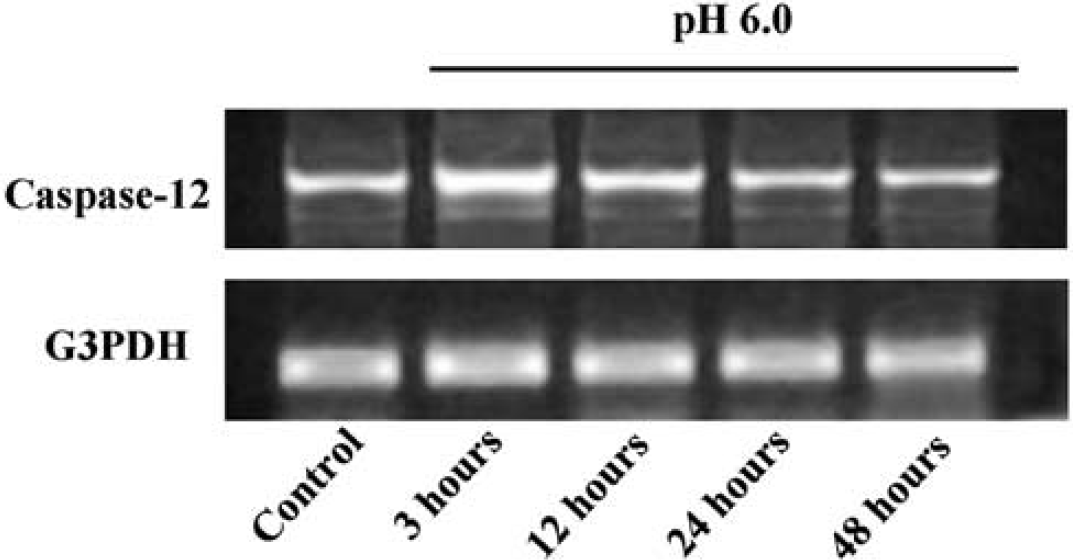
Caspase-12 mRNA expression after acidosis. Bands show products of caspase-12 mRNA RT-PCR from samples prepared from astrocyte cultures at serial time points after 3 hours incubation in pH 6.0 medium. Reverse transcription-polymerase chain reaction of G3PDH mRNA served as a control at each time point. Representative of four studies with similar results.
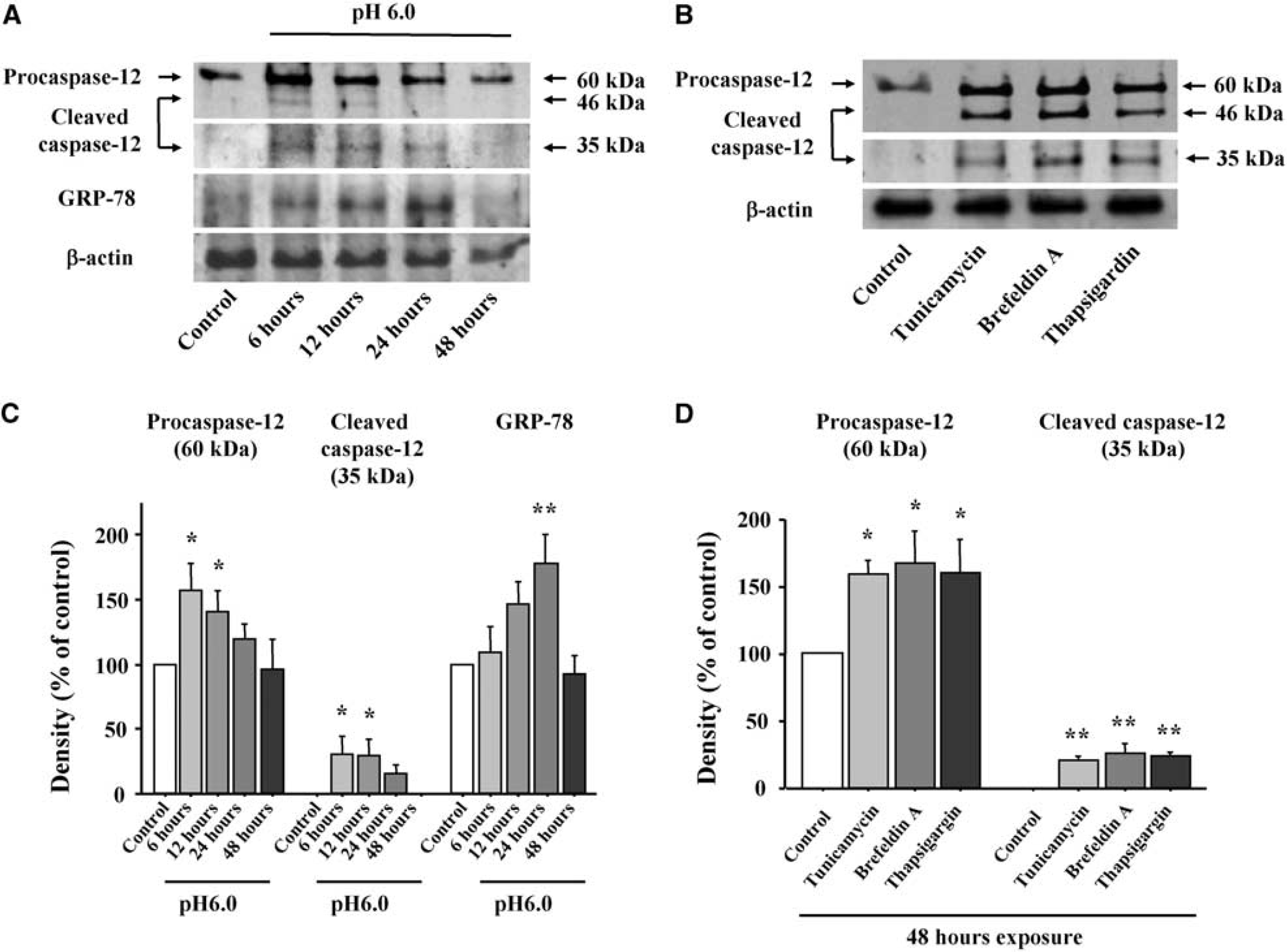
Expression of GRP-78, procaspase-12 and cleaved caspase-12 after acidosis and ER stress. Western blots were prepared from astrocytes at designated time points after (
Activated caspase-12 can activate caspase-9 (Morishima et al, 2002; Rao et al, 2002a), which can in turn induce caspase-3 activation and cell death. As shown in Figure 4, the pan-caspase inhibitor zVAD-fmk reduced astrocyte death induced by both acidosis (A) and tunicamycin (B), suggesting that caspase activation contributes to astrocyte death after acidosis and ER stress. The reduced astrocyte death observed in cultures treated with cycloheximide further suggests that de novo protein synthesis contributes to this cell death process.
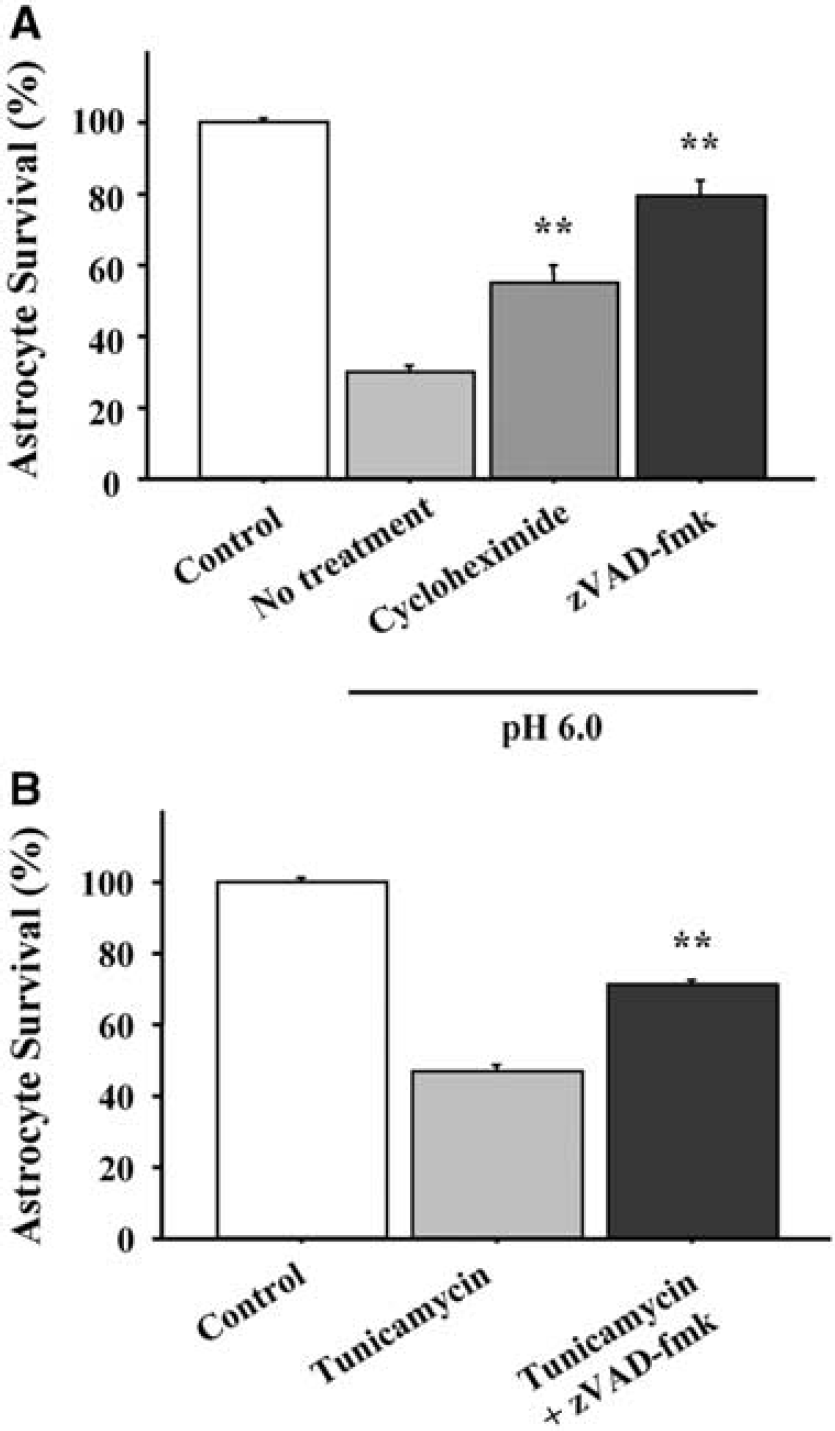
Protein synthesis and caspase inhibitors reduce astrocyte death after acidosis and ER stress. (
Antisense Downregulation of Caspase-12 Reduces Acidosis-Induced Cell Death
To test for a causative role of caspase-12 in acidosis-induced astrocyte death, we used antisense PMOs to inhibit the upregulation of caspase-12 induced by acidosis. The time course of the antisense PMO treatment, acidosis exposures, and cell harvesting are diagrammed in Figure 5A. Antisense PMO treatment of cultures that were not exposed to acidosis reduced the expression of the 60 kDa pro-form of caspase-12 (Figure 5B), indicating significant turnover of caspase-12 over the 18-hour interval between antisense treatment and cell harvest. Antisense PMO treatment 6 hours before acidosis reduced the acidosis-induced upregulation of the 60 kDa pro-form of caspase-12 by 63% (P<0.05, n=3) and reduced the 35 kDa active caspase-12 fragment to nearly undetectable levels (Figure 5B). As shown in Figure 5C, caspase-12 antisense PMO treatment 6 hours before acidosis also reduced the acidosis-induced astrocyte cell death, resulting in a greater than 2-fold increase in cell survival relative to cultures treated with the control, random-sense oligonucleotide. DAPI staining showed a corresponding decreased number of apoptotic cells in caspase-12 antisense-treated cultures (Figure 5D).
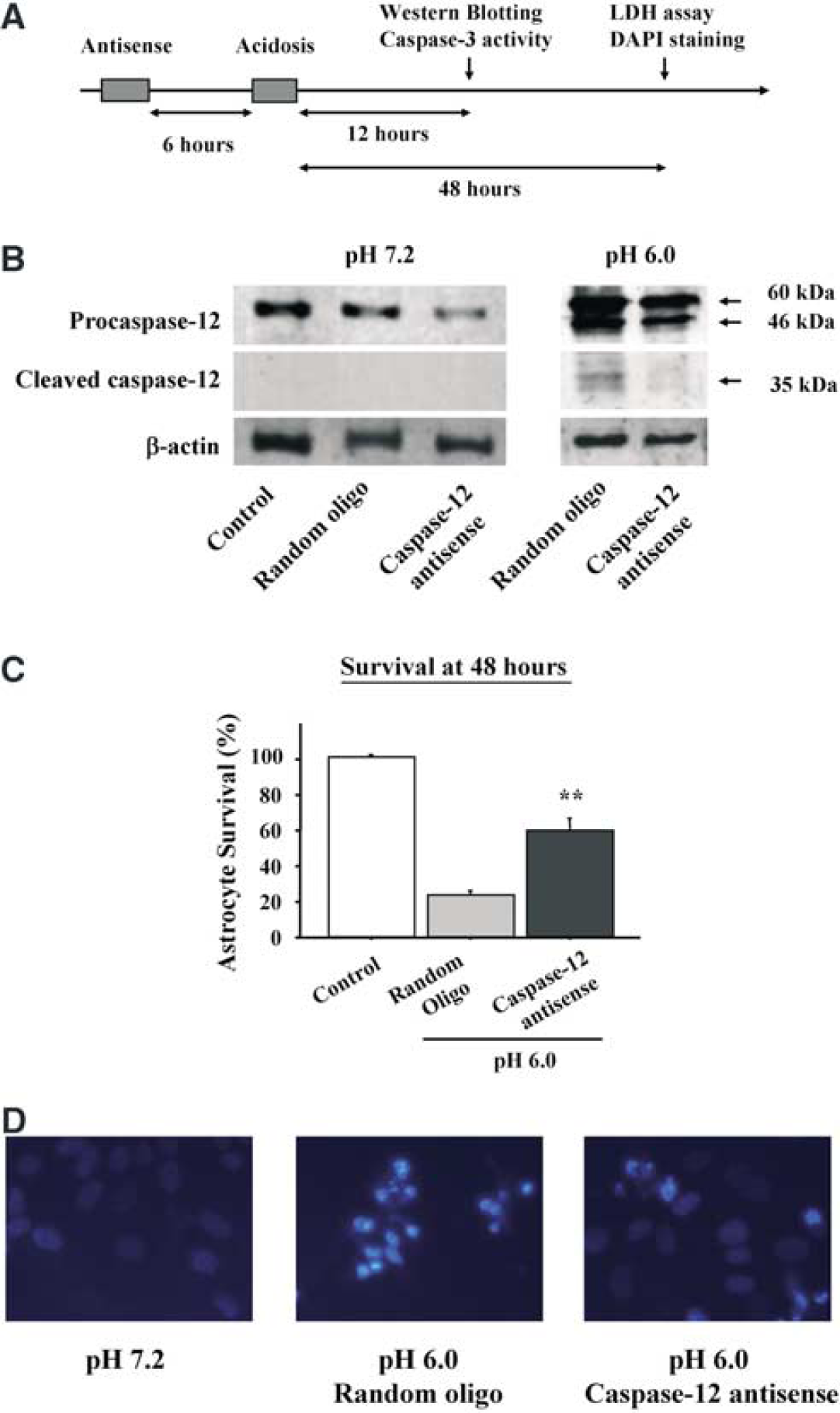
Effect of antisense downregulation of caspase-12. (
The residual caspase-12 in the antisense-treated cultures (Figure 5B) can be attributed in part to caspase-12 protein present in the cells before antisense treatment. Antisense treatment performed 24 hours before acidosis substantially reduced the basal caspase-12 protein expression, similar to that shown in Figure 5B at pH 7.2, but had only modest effect on the acidosis-induced upregulation of caspase-12 expression (not shown). Treatment with antisense PMO twice, at both 24 and 6 hours before acidosis, caused substantial cytotoxicity. Accordingly, all subsequent antisense studies were performed with antisense PMOs added 6 hours before acidosis, recognizing that this produces an incomplete suppression of caspase-12 expression.
Acidosis-Induced Caspase-12 Activation is Linked to Caspase-3 Activation
Since caspase-12 activation can lead to cell death by activation of the downstream, effector caspase-3 (Kuida et al, 1998; Morishima et al, 2002), we measured the activity of caspase-3 in acidosis-treated cultures. As shown in Figure 6, caspase-3 activity was substantially elevated in cell lysates harvested between 6 and 48 hours after acidosis, with a peak observed at 24 hours (Figure 6A). This increase was reduced by roughly 40% in sister culture wells that had been treated with caspase-12 antisense oligonucleotides 6 hours before acidosis (Figure 6B).
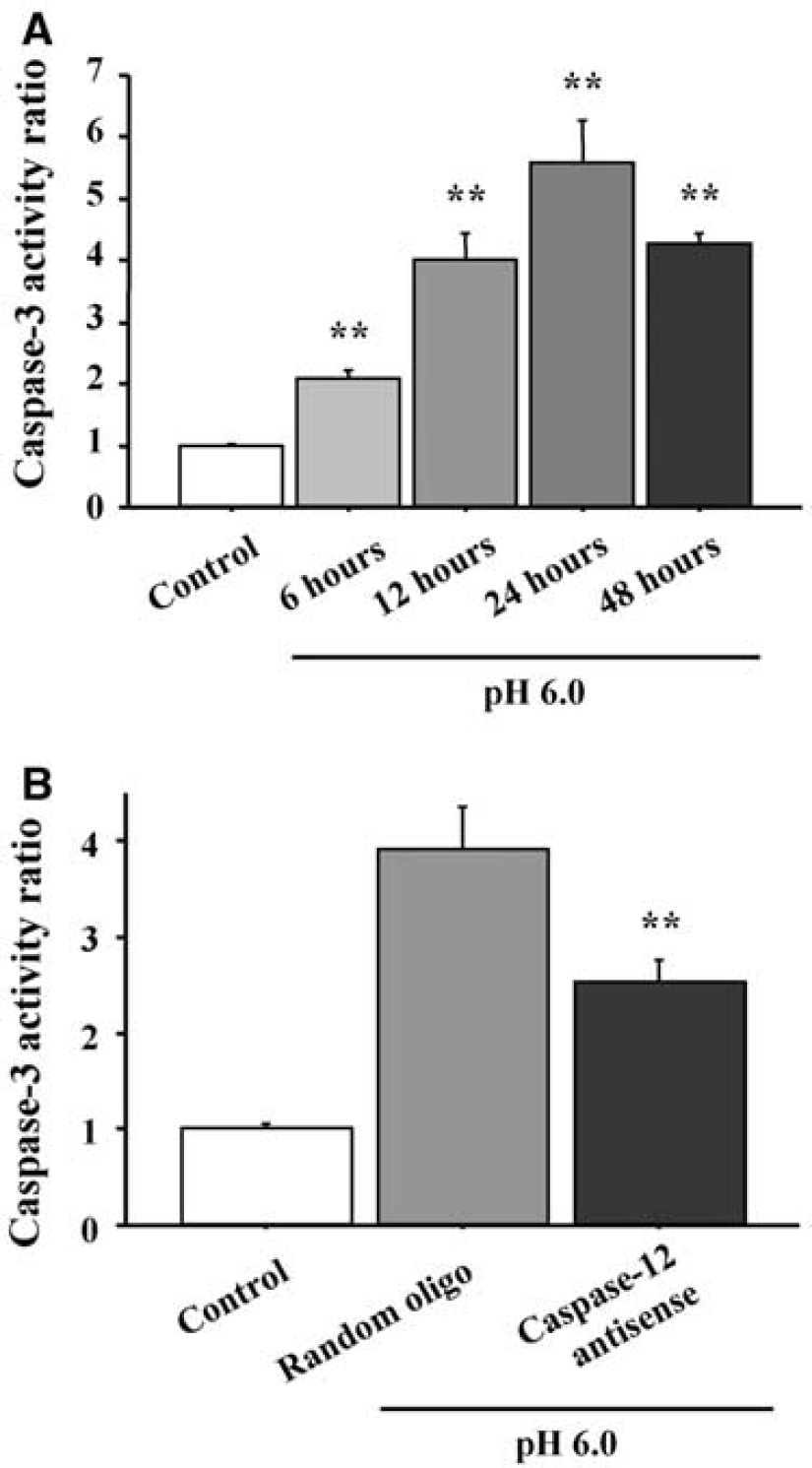
Effects of acidosis and caspase-12 antisense oligonucleotides on caspase-3 activity. (
Acidosis Induces Dissociation of Ire1-α and Procaspase-12 from GRP-78
Normally, Ire1-α and procaspase-12 are both bound to GRP-78 on ER membranes (Bertolotti et al, 2000; Rao et al, 2002b). During ER stress, GRP-78 binds to misfolded proteins in the ER lumen and releases Ire1-α and procaspase-12, which together lead to the cellular ER stress response. To determine whether acidosis may induce the ER stress response by this mechanism, immunoprecipitation was performed with anti-GRP-78 antibody, and the immunoprecipitates were analyzed with Western blots probed with antibodies to Ire1-α and caspase-12. As shown in Figure 7, cultures lysed at either 6 or 12 hours after acidosis showed reduced binding of both Ire1-α and procaspase-12 to GRP-78. Of note, the decrease in procaspase-12 binding was observed despite an increase in total procaspase-12 at these time points (shown in Figure 3).
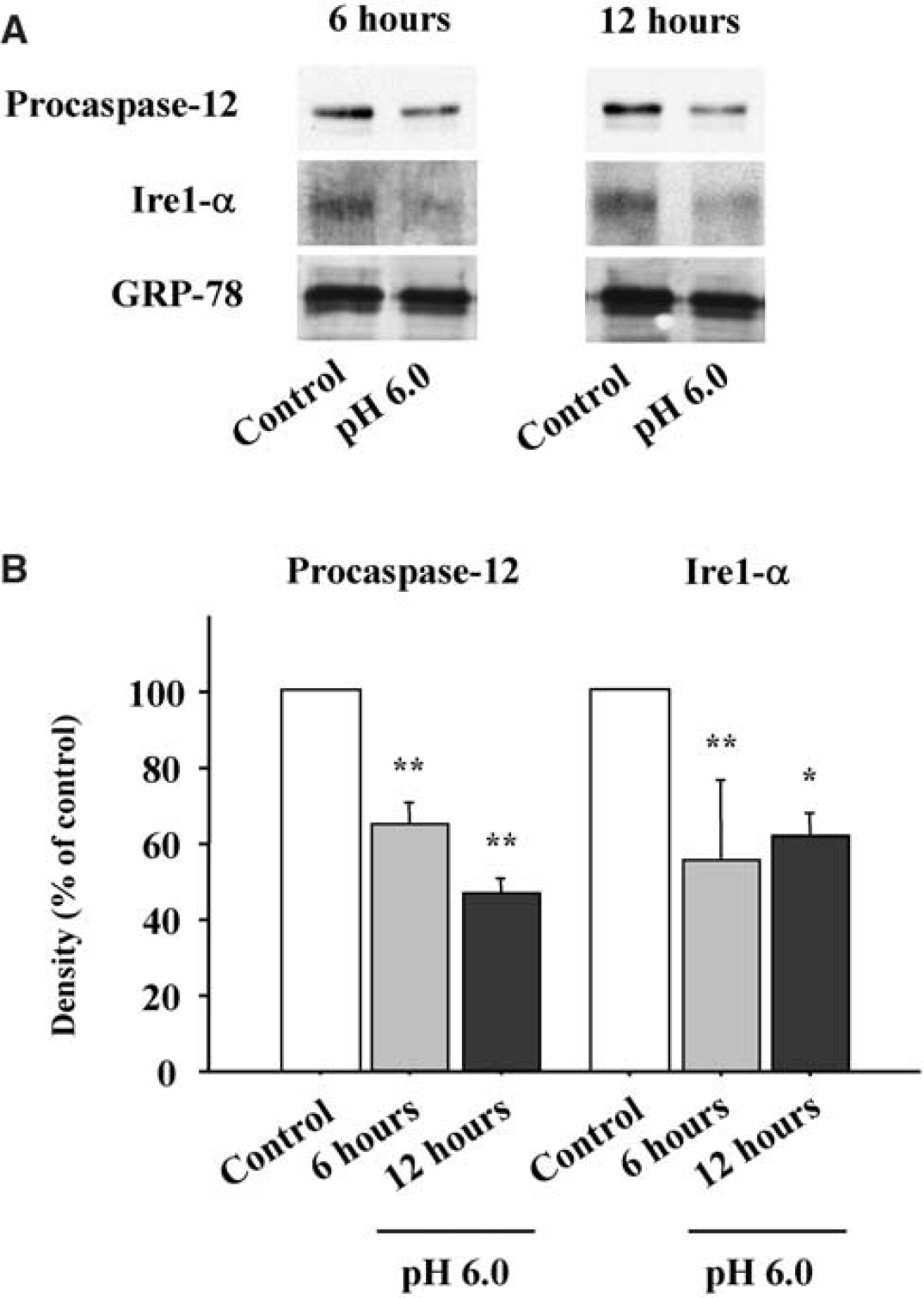
Caspase-12 and Ire1-α Western blots from GRP-78 immunoprecipitates. Samples were collected at 6 and 12 hours after incubation at pH 6.0 for 3 hours. (
Transient Focal Ischemia in Rat Brain Induces Caspase-12 Expression in Peri-Infarct Astrocytes and Neurons
Immunostaining for caspase-12 was performed on rat brains subjected to transient focal ischemia to identify the regions and cell types in which caspase-12 expression is induced (Figure 8). The area of infarction was verified by hematoxylin and eosin staining (Figure 8C). Caspase-12 was upregulated specifically in the periinfarct region, which contains viable cells that have been exposed to acidosis (Obrenovitch, 1995). Increased GFAP expression was also observed in this region, indicative of reactive astrocytosis. Confocal images confirmed that the GFAP-positive astrocytes were a primary locus of both caspase-12 and GRP-78 protein expression (Figures 8D and 8E), although increased caspase-12 expression also colocalized with MAP-2, a cell type marker for neurons (Figure 8F). Almost all of the astrocytes in the periinfarct region were strongly immunopositive for caspase-12. Of these, 10% to 30% showed nuclear condensation indicative of apoptosis (Figure 8G), with the frequency of nuclear condensation increasing with proximity to the infarct border.
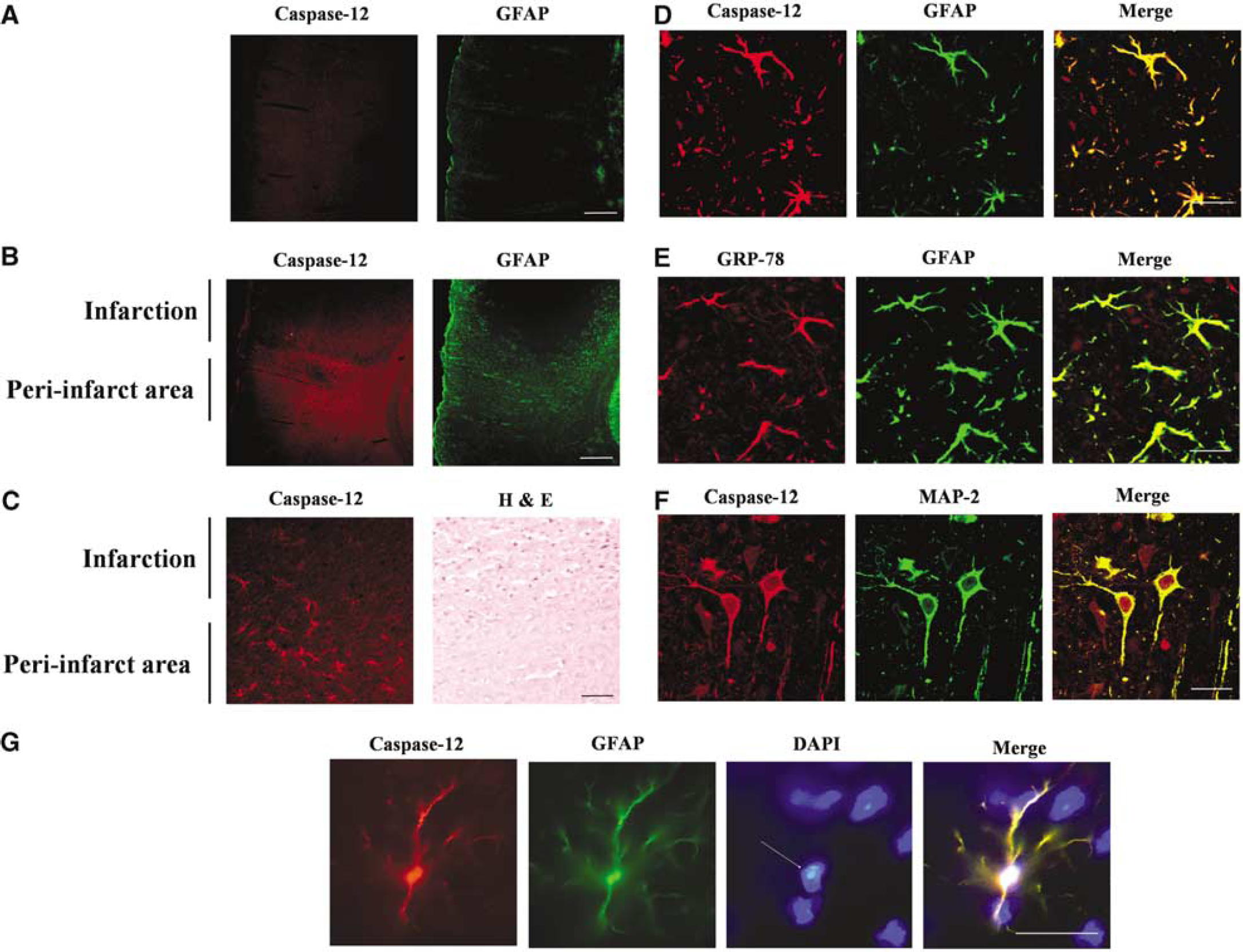
Endoplasmic reticulum stress protein expression in post-ischemic brain. (
Discussion
Acidosis has not previously been recognized as a factor inducing ER stress. The present studies show that acidosis in astrocytes causes upregulation of the ER stress proteins GRP-78 and caspase-12, along with cleavage of caspase-12 to its active fragments. These findings also suggest a contribution of the ER stress pathway to delayed astrocyte death after acidosis, since antisense downregulation of caspase-12 reduced both caspase-3 activation and astrocyte death. The dissociation of Ire1-α and procaspase-12 from GRP-78 after acidosis suggests this as the mechanism by which the ER stress response is triggered by acidosis.
Caspase-12 normally exists in an inactive, 60 kDa procaspase form that is bound to ER membranes in association with GRP-78 (Rao et al, 2002b). During ER stress, caspase-12 dissociates from the ER membrane and is cleaved first to a 46 kDa fragment and subsequently to an active, 35 kDa fragment (Nakagawa and Yuan, 2000; Nakagawa et al, 2000; Rao et al, 2002b). The expression levels of these caspase-12 species are consequently influenced by the rates of both de novo procaspase-12 synthesis and procaspase-12 cleavage. Measures of caspase-12 mRNA presented here showed a roughly two-fold increase after acidosis, followed by increased expression of both full-length procaspase-12 and its cleavage products. The magnitude of these increases is similar to that previously reported in vivo after brain ischemia and trauma (Harrison et al, 2001; Larner et al, 2004). A pattern of caspase-12 expression similar to that produced by acidosis was also induced by the ER stress agents thapsigargin, tunicamycin, and brefeldin A. The concentrations of ER stress agents used for these studies were sublethal to the astrocytes, though higher than typically required for induction of ER stress in other cell types (Nakagawa et al, 2000; Rao et al, 2001; Fujita et al, 2002).
The reduction in acidosis-induced astrocyte death by the broad-spectrum caspase inhibitor zVAD-fmk suggested caspase involvement in the cell death process. Biochemical measurements of caspase-3 activity after acidosis confirmed an increase in the activity of this effector caspase between 6 and 48 hours after acidosis, a time course that roughly corresponds to the time course of caspase-12 activation. Caspase-12 can promote caspase-3 activation by cleavage activation of caspase-9 (Morishima et al, 2002; Rao et al, 2002a). Consistent with this, antisense downregulation of caspase-12 reduced peak caspase-3 activity after acidosis and reduced acidosis-induced astrocyte death.
Endoplasmic reticulum stress is induced by an excess of misfolded proteins in the ER lumen. There are several possible ways by which acidosis could affect the folding of ER proteins, including direct denaturation, inhibition or inactivation of enzymes involved in ER protein processing, and inhibition of the ER Ca2+/ATP-ase (Wolosker et al, 1997). Misfolded proteins in the ER induce release of Ire1-α and caspase-12 from the ER chaperone protein GRP-78 (Bertolotti et al, 2000; Rao et al, 2002b). There are two mammalian Ire1 proteins, Ire1-α and -β, and both participate in ER stress signaling. Ire1-α is broadly expressed, whereas Ire1-β is expressed selectively in foregut-derived epithelium (Tirasophon et al, 1998; Wang et al, 1998). Results of the present study demonstrated a dissociation of Ire1-α from GRP-78 after acidosis, suggesting this as a likely mechanism by which the ER stress response is induced by acidosis. Of note, Ire1-α may also be involved in caspase-12 activation. Activated Ire1-α can bind to tumor necrosis factor receptor associated factor 2 (TRAF2), and the Ire1-α/TRAF2 complex interacts with procaspase-12 to promote its clustering and activation (Urano et al, 2000; Yoneda et al, 2001).
Immunostaining of brain sections after transient middle cerebral artery occlusion was performed to determine whether an ER stress response could be identified in astrocytes in vivo after ischemia. These studies showed marked upregulation of caspase-12 in astrocytes in the periinfarct region. Caspase-12 upregulation was highly correlated with GRP-78 upregulation, suggesting that both occur as aspects of the ER stress response in these cells. Some of these cells were seen to have nuclear condensation consistent with apoptotic astrocyte death. The nuclear condensation changes are similar to those observed in the cell culture studies presented here and consistent with prior reports of delayed apoptotic changes in astrocytes after ischemia–reperfusion (Chen et al, 1997; Krupinski et al, 2000; Takuma et al, 2004). These findings are consistent with a role for acidosis and ER stress in delayed astrocyte death after ischemia, but do not exclude the possibility that factors other than acidosis may trigger or contribute to these changes in the periinfarct astrocytes.
Acidosis has long been recognized as a major factor contributing to cell death after cerebral ischemia (Rehncrona et al, 1981; Plum, 1983; Siesjo, 1988; Nedergaard et al, 1991). More recently, ER stress has also been recognized as an important mediator of ischemic cell death (Paschen and Frandsen, 2001; DeGracia et al, 2002). The present studies identify a novel link between these two factors, and the dissociation of Ire1-α and procaspase-12 from GRP-78 induced by acidosis suggests a likely mechanism by which ER stress is triggered by acidosis. Since the signaling pathways and machinery of the ER stress response are similar among mammalian cell types (DeGracia et al, 2002; Harding and Ron, 2002), the link between acidosis and ER stress is also likely to hold true in other cell types. Of note in this regard, colabeling with cell type markers showed the upregulation of caspase-12 in neurons as well as in astrocytes in the periinfarct region, consistent with prior reports that focused on neuronal caspase-12 expression (Mouw et al, 2003; Shibata et al, 2003).
The condition most commonly employed in studies of ischemic cell death in vitro is combined oxygen and glucose deprivation, without superimposed acidosis. An advantage to the cell culture approach in general is that the several metabolic disturbances resulting from ischemia can be independently controlled to identify discrete mechanisms involved in the cell death process. However, models of cerebral ischemia that do not include acidosis might be overly simplistic. The results of the present study and many others (Giffard et al, 1990b; Tombaugh and Sapolsky, 1990; Swanson et al, 1995, 1997; Sapolsky et al, 1996; Ying et al, 1999; Bondarenko and Chesler, 2001) suggest that cellular responses to ischemic conditions are highly influenced by changes in pH.
By the same token, a limitation to the cell culture studies presented here is that acidosis is studied in isolation. Ischemia produces, in addition to acidosis, changes in extracellular ion composition, release of neuroactive substances, production of reactive oxygen species, oxygen deprivation, and variable degrees of glucose deprivation (Peek et al, 1989; Obrenovitch, 1995; Sharp et al, 1998). Each of these factors could influence the link between acidosis and the ER stress response, or contribute independently to the ER stress response. In particular, reactive oxygen species can cause protein denaturation (Stadtman and Levine, 2003) and could thereby trigger ER stress by mechanisms similar to those of acidosis. Thus, the present results indicate that acidosis could be a significant factor contributing to ER stress-mediated astrocyte death after cerebral ischemia, but additional work will be required to establish the quantitative significance of this effect in vivo.
Footnotes
Acknowledgements
We thank Dr Stephen Massa (UCSF), and Dr Rammohan Rao (Buck Institute for Age Research) for helpful discussions. This work was supported by the REAP and Merit Review programs of the US Department of Veterans Affairs.