
Abstract
Integrins play critical roles in the process of angiogenesis and are attractive targets for anticancer therapies. It is desirable to develop new types of small-molecule inhibitors of integrin. Herein, the binding features of several inhibitors to integrin αvβ3 have been studied by surface plasmon resonance (SPR) biosensor technology and molecular docking analyses. The SPR results indicated that the equilibrium dissociation constant (KD) values are evaluated for the inhibitors and showed that the KD value of cyclopeptide c-Lys is much lower than the reference molecule. In addition, the 3D structural model of integrin αvβ3 was generated according to the crystal structure of the integrin αvβ3 complex, and the molecular docking simulation analyses revealed that the predicted binding sites for the most active cyclopeptide c-Lys were consistent with the reported structure. These results thus implied that cyclopeptide c-Lys could be developed as a novel inhibitor for integrin αvβ3. The current work has potential for application in structure-based integrin αvβ3 inhibitor discovery.
Introduction
I
Recently, the surface plasmon resonance (SPR) biosensor has been recognized as a valuable tool in monitoring biological interactions with advantages of being label-free, real-time, noninvasive measurements; having low sample consumption; and being widely used in hit determination. 9 In our study, the SPR technology was successfully applied to determine the binding affinities of several cyclopeptides to integrin αvβ3. The equilibrium dissociation constant (KD) values evaluated by the Biacore X100 for the inhibitors demonstrated that the KD value of cyclopeptide c-Lys is much lower than that of the reference inhibitor.
To further investigate the inhibitor/integrin αvβ3 binding mode at the atomic level, the LigandFit method of molecular docking was employed to dock the newly designed molecules in the active site of integrin αvβ3. Molecular docking has become a widely used technology in drug design to identify and optimize lead compounds. 10,11 During the past few years, some molecular docking algorithms based on different search methods have been developed and implemented. 10,12 Molecular docking involves the prediction of a ligand (small-molecule) conformation and orientation within the active site of the molecular target. The molecular docking program LigandFit, 13 a shape-based method for docking ligands into protein active sites, was used to perform the virtual screening. The aim was to build models able to (1) distinguish active from inactive compounds for integrin receptor αvβ3, (2) predict binding affinity for active compounds, and (3) find selective compounds. In the current work, molecular docking was performed with the 3D structural model of integrin αvβ3 generated on the basis of the crystal structure of the integrin αvβ3-ligand complex (PDB entry code 1L5G). 14 The predicted binding sites for the most active cyclopeptide c-Lys were consistent with the reference structure, but the inactive cyclopeptide c-Leu was not, which suggests that cyclopeptide c-Lys could be developed as a potential inhibitor for integrin αvβ3.
In our previous work, a new family of compounds (17 cyclopeptides) was prepared according to the published procedure, 15 as shown in Figure 1 . In the current work, the binding features of several inhibitors to integrin αvβ3 have been studied by SPR technology and molecular docking simulation.
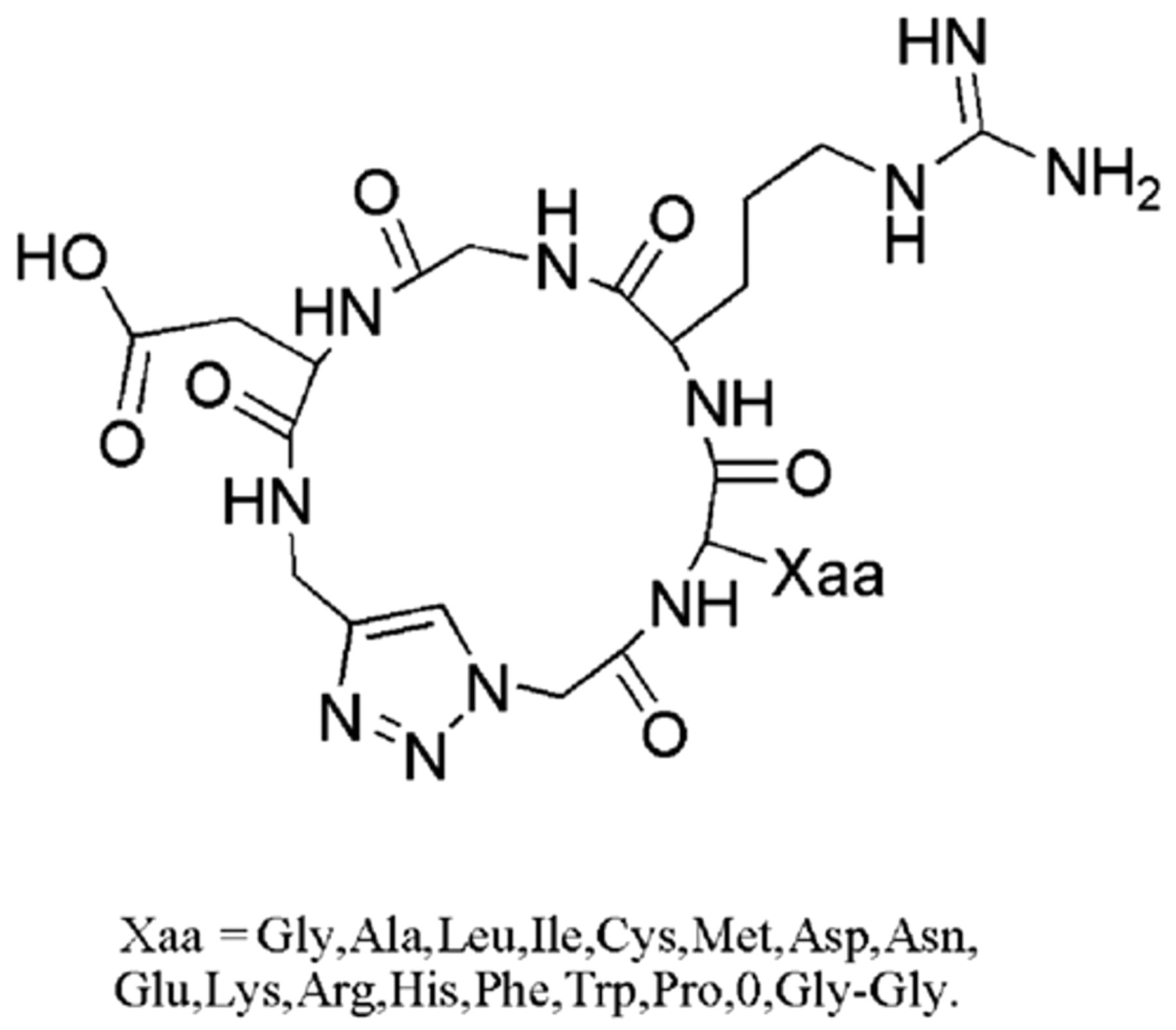
Structure of cyclo[-Arg-Gly-Asp-ψ(triazole)-Gly-Xaa-].
Materials and Methods
Materials
Protein integrin αvβ3 was purchased from Millipore (Billerica, MA). N-octyl-β-D-glucoside was purchased from Dojindo Laboratories (Kunamoto, Japan). The cyclopeptides cyclo[-Arg-Gly-Asp-ψ(triazole)-Gly-Xaa-] (c-Xaa) ligands for integrin αvβ3 receptor as previously described 15 were synthesized for surface plasmon resonance experiments. N-hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethyl aminopropyl) carbodiimide hydrochloride (EDC) were obtained from JK Chemical (Beijing, China). All other chemicals were of reagent grade or ultrapure quality and purchased from Shanghai Sangon Biological Engineering & Technology and Service Co. Ltd. (Shanghai, China).
Methods
SPR technology-based binding assay
To inspect the possible active compounds with binding affinities to integrin αvβ3, SPR technology-based Biacore X100 biosensor was used (GE Healthcare, Piscataway, NJ). The SPR experiment was performed according to the manufacturer’s instruction. In brief, integrin αvβ3 was immobilized on a CM5 chip (Biacore, Piscataway, NJ) by the amine-coupling method. The dextran surface was activated by injection of NHS/EDC mixture (0.1M NHS and 0.4M EDC, 1:1 mixture) for 7 min. Integrin (in 10 mM acetate, pH 4.0) was injected over activated surface. Finally, unreacted activated groups were blocked by a 35-µL injection of ethanolamine. All the binding experiments were performed at 25°C at a continuous flow rate with 10 mM HEPES buffered saline (10 mM HEPES, 150 mM NaCl, 1 mM MgCl2, 1 mM MnCl2, and 5 mM n-octyl-β-D-glucoside, pH 7.4). A few sets of experiments, using cyclopeptides as analytes, were run on the chip surface at the concentration of 0, 3.2, 6.3, 12.5, 25, 50, and 100 µM. The cyclopeptide cRGDfK, as a positive control, which displayed a high binding affinity to integrin αvβ3, was reported previously. 16 The sensor surfaces were regenerated with a short pulse of 10 mM glycine, pH 2.2.
All the equilibrium dissociation constants (KDs) used for evaluating the ligand-integrin αvβ3 binding affinity were determined with Biacore evaluation Version 4.1 software (Biacore). During the assays, the signal was corrected against the control surface response to eliminate refractive index changes due to buffer change.
SPR data analysis
During the assays of integrin αvβ3 and its inhibitors with Biacore X100, the binding response signals in RUs were continuously recorded graphically as a function of time. The data were fit using a homogeneous single-site interaction model, A + B = AB, where A represents the injected analyte, B is the immobilized ligand, and AB is the complex formed during the reaction. The Biacore results indicated that, in reaching equilibrium, both the association with and dissociation from the immobilized integrin αvβ3 of the analytes involved slow phases, as shown in Figure 2 (using c-Lys as a typical example). Therefore, the data were analyzed by global fitting to a 1:1 binding model (Langmuir) of both the association and dissociation phases for several concentrations simultaneously, using the BIAevaluation 4.1 software. 17 The curve-fitting efficiency was evaluated by chi-square. The chi-square value denotes the fitting degree between the estimative and the experimental curves.
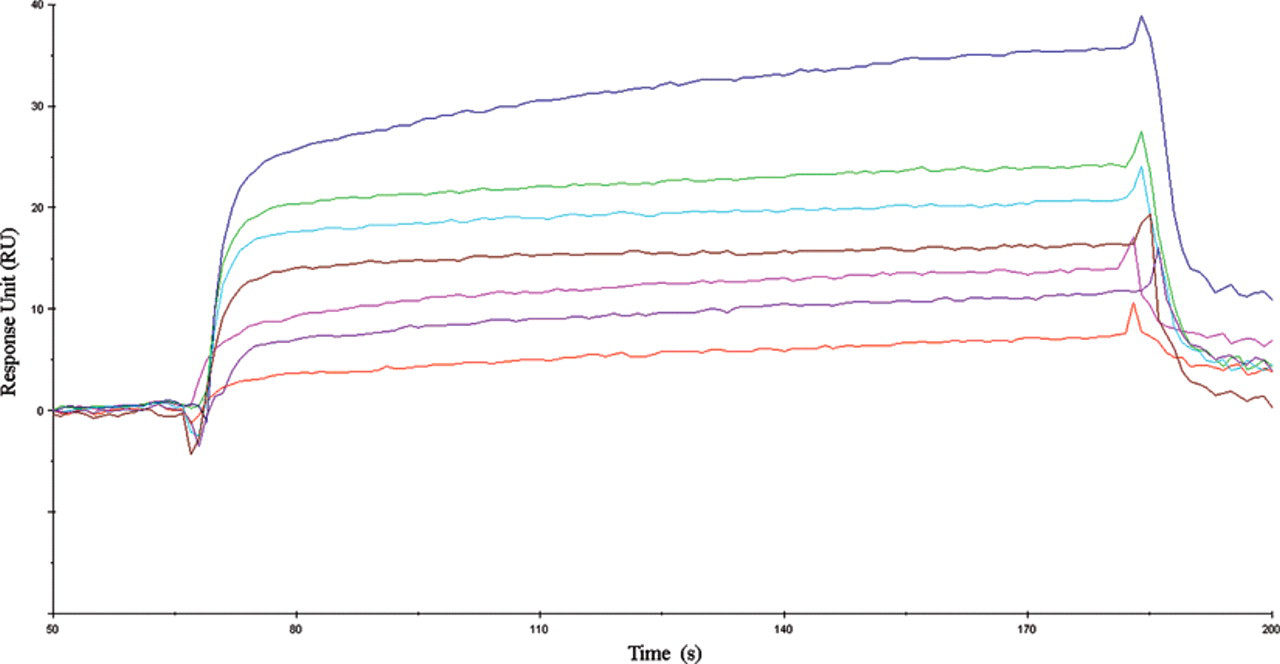
Kinetic analysis of the cyclopeptide c-Lys binding to integrin αvβ3 by surface plasmon resonance (SPR) technology. Representative sensorgram obtained from injection of cyclopeptide c-Lys at concentrations of 0, 3.2, 6.3, 12.5, 25, 50, and 100 µM.
Molecular modeling systems
The X-ray structure of integrin αvβ3 was obtained from the PDB (1L5G, resolution 3.20 Å). 14 The structure of αvβ3, a heterodimeric protein (183 kDa) of 1649 amino acids, consists of noncovalently linked alpha V subunit polypeptide (CD51) and beta 3 subunit polypeptide (CD61). The 3D-optimized structures of cyclopeptides were generated by the program ChemSketch/3D Viewer (Version 11.0, from Advanced Chemistry Developments, Toronto, Canada; http://www.acdlabs.com). LigandFit was used for molecular modeling on a Linux workstation. The resultant structures were then analyzed using the LigPlot 4.22 18 program to identify specific contacts between ligands and the protein. The visualization of complexes was employed by the Pymol 0.99 program 19 on a Windows XP workstation.
LigandFit docking procedure
LigandFit, a docking tool, was used to dock ligands in the protein binding site, calculate scores, and rank them accordingly (http://www.accelrys.com/). The docking site was defined using the LigandFit site search utility. Two possibilities for binding site definition were offered by LigandFit: cavities (i.e., areas of unoccupied grid points) can be detected based on the shape of the protein or from a bound ligand. 20 The former strategy has to be applied when only the apo (ligand-free) structure of the protein is known, which involves more uncertainty of the obtained results, but the latter strategy is more reliable and the technique of choice for holo (ligand-bound) protein structures. In our case, the binding site was generated from the X-ray pose of the bound ligand. The grid resolution was set to 0.5 Å, and the ligand-accessible grid was defined such that the minimum distance between a grid point and the protein was 2.0 Å for hydrogen and 2.5 Å for heavy atoms. Such a general binding site was used to calculate the nonbonded interactions between all atoms of each ligand and the protein residues. If necessary, the initial binding site cavity was expanded with the program expansion tool to a distance of 3 Å in all directions and then edited manually to remove points protruding far outside the molecular surface. After characterization of the binding pockets, a Monte Carlo method was employed for the conformational search of the ligand. Ligands were docked within the identified site with 100 possible conformations, with each ranked according to the dock score provided in LigandFit. Dock scoring was performed with LigScore1 scoring function. 13
Results and Discussion
SPR-based binding assays between cyclopeptides and the integrin
The interactions of the cyclopeptides were investigated with the immobilized integrin αvβ3 on the CM5 sensor chip at a level of 11,000 response units (RU) with the standard amine coupling procedure. Subsequently, the chip was used to measure binding parameters for the interaction with different cyclopeptides (analytes) at concentrations ranging from 3.2 to 100 µM. As shown in Figure 2 , the RU values evaluating the cyclopeptide c-Lys binding to the integrin αvβ3 revealed an obvious concentration-dependent manner, which demonstrated that the determined equilibrium dissociation constant (KD) of 1.3 × 10−9 M is much lower than the KD value of 1.3 × 10−6 M for the reference molecule cRGDfK. 16 The binding kinetic constant was fitted using the 1:1 binding fitting model (Langmuir) supplied in the Biacore software. Using this model, the chi-squared value of c-Lys was calculated to be 0.79, indicating a good fit.
Shown in Table 1 are the KD values of all the tested inhibitors binding to integrin αvβ3 from the SPR assay. In addition to the c-Lys mentioned above, for c-Gly, it is nearly within the same quantitative grade (2.4 × 10−6 M) compared with the reference molecule, and for other cyclopeptides, their KD values (>10−6 M) are higher. The chi-squared value was used to judge the quality of fit between experimental data and that derived from binding models. The smaller the chi-square, the smaller their variation. Typically, good fittings derive chi-square values less than 10. 21 All the results are summarized in Table 1 . To sum up, we compared the KD values of all compounds obtained from the SPR assay, and the KD value of c-Lys was much lower than the reference molecule.
Equilibrium Dissociation Constant (KD) Values of Cyclopeptides Determined by the SPR Assay
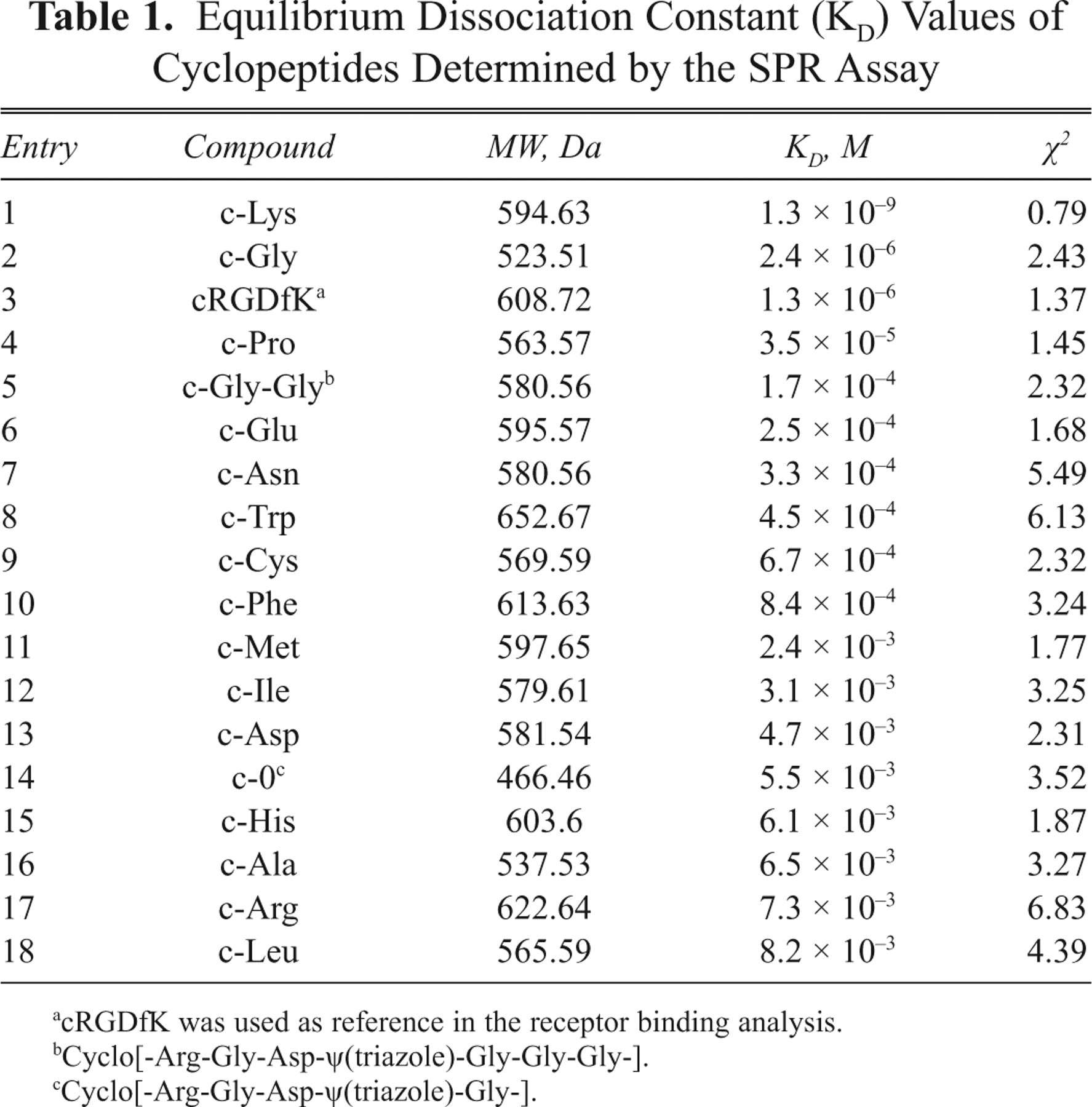
cRGDfK was used as reference in the receptor binding analysis.
Cyclo[-Arg-Gly-Asp-ψ(triazole)-Gly-Gly-Gly-].
Cyclo[-Arg-Gly-Asp-ψ(triazole)-Gly-].
Model evaluation of LigandFit docking
To explore the binding characteristics of the inhibitors to integrin αvβ3 at the molecular level, molecular docking was applied to construct the inhibitor/integrin αvβ3 binding model. The LigandFit program was employed to predict the potential binding of cyclopeptides to integrin αvβ3 based on the cavity detection algorithm. In our case, the binding site was generated from the X-ray pose of the bound ligand. Previous crystallographic and modeling analysis showed that cRGDfNMeV interacts with integrin αvβ3 mainly using the R-G-D moiety. 14 Evaluation of the site definition was achieved by superimposition of the docked and the X-ray conformations and orientations of cRGDfNMeV, as shown in Figure 3 . The root mean square deviation (RMSD) of the best docked structure from the X-ray form was 0.234 Å. This promising result confirmed the applicability of our binding site model for further studies.
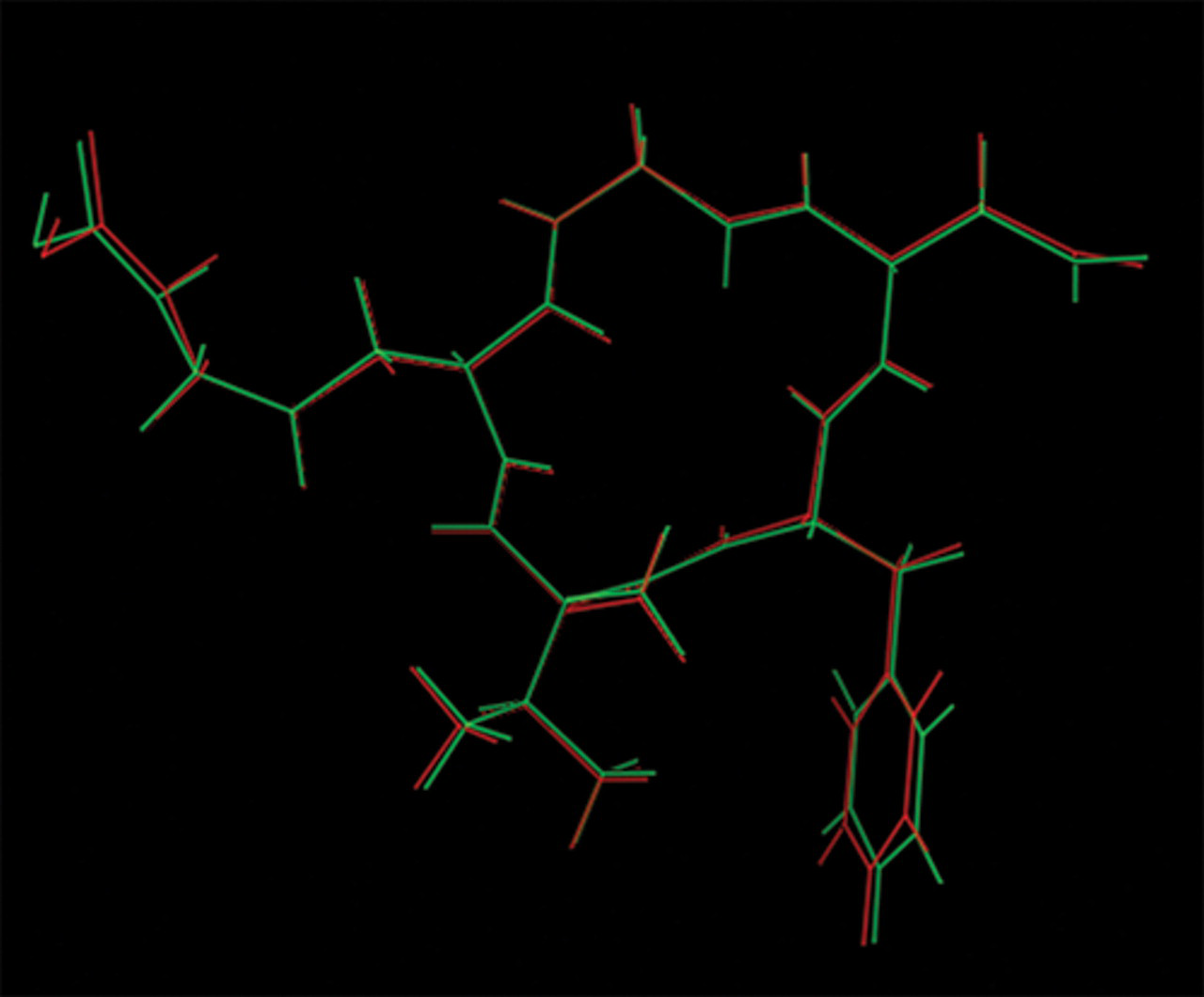
Superimposition of the docked conformation (red) with the X-ray conformation (green) of the ligand cRGDf NMeV (PDB entry 1L5G).
Analysis of docking complexes
The docked conformations were scored using different scoring functions (LigScore1, LigScore2, PLP1, PLP2, JAIN, and PMF). Searching the lists of scoring functions for the cyclopeptides showed that the actives obtained highest positions, when ranked according to their LigScore1 values. Hence, it can be stated that the good performance of LigScore1 justifies the use of this scoring function to rank the results of a LigandFit docking process for this system. Three descriptors are used to calculate the LigScore (a program-internal scoring function): a softened Lennard-Jones potential, 1 count of the buried polar surface area between a protein and ligand that involves attractive protein-ligand interactions, and the sum of squared attractive and repulsive protein-ligand contact terms. The 3-descriptor system that could be found is equation (1):
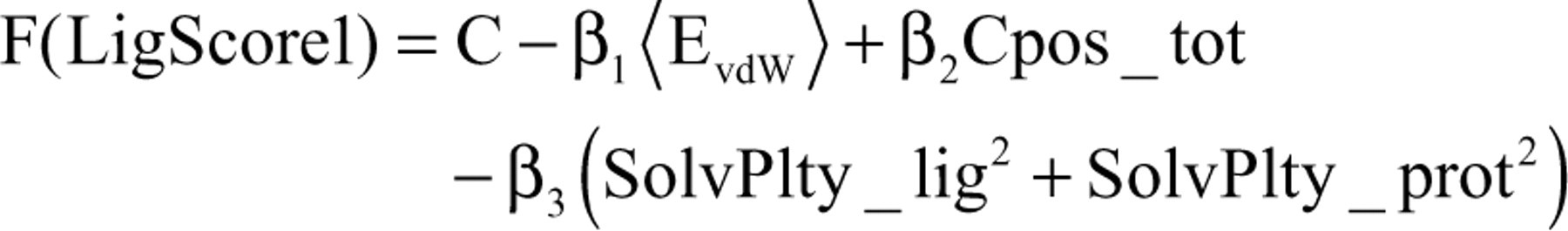
where EvdW indicates the van der Waals (vdW) energy of protein-ligand interactions, which is computed via a Lennard-Jones potential 13 using van der Waals radii and energy parameters of either the consistent force field (CFF) or the DREIDING force field. Cpos_tot is the total surface area of the ligand involved in attractive polar interactions with the protein. The SolvPlty_lig2 term represents the overall polar ligand surface that is buried inside the protein binding site, and the SolvPlty_prot2 term indirectly accounts for the polar protein surface in contact with the bound ligand molecule. Therefore, in the case of cyclopeptide/integrin αvβ3 complexes, the contributions of 3 key descriptors (vdW and 2 surface descriptors) may be identified effectively.
Take the example of c-Lys, the most active compound in SPR assay. Molecular docking results thus describe the general binding mode of the inhibitor c-Lys to integrin αvβ3. The hydrophobic and hydrogen bond interactions between integrin αvβ3 and the various cyclopeptide ligands were visualized by employing the program LigPlot. The schematic binding mode analysis of c-Lys/integrin αvβ3 complex shows the residues involved in the receptor site shown in Figure 4 . On the whole, c-Lys has possible strong hydrogen bonds with 9 amino acid residues—Asp150(A), Tyr178(A), Asp218(A), Asp219(A), Ser123(B), Asn 215(B), Ala218(B), Glu220(B), and Arg241(B)—of the αvβ3 receptor, as judged by the LigPlot program. Four residues of them—Asp150(A), Tyr178(A), Asp218(A), and Asn215(B)—are in good agreement with those usually observed for other complexes in the literature. 14 Thus, new hydrogen bonds have been found in the complex. The presence of additional hydrogen bonds can stabilize the complex structure but could contribute to the R-G-D moiety conformation. Moreover, analysis with the LigPlot program suggests that c-Lys can form 3 hydrophobic interactions with Tyr178(A), Ala218(B), and Tyr122(B) of the αvβ3 receptor. The docking conformation of c-Lys around the receptor binding domain of integrin αvβ3 is depicted in Figure 5 . The Asp residue adopted a U-shape with the 2 Arg residues in the docking conformation of c-Lys. However, there is a strong salt bridge between the Asp carboxylate oxygens of c-Lys and the Mn4001(B) ion, which is also involved in hydrogen bond interactions with the hydroxyl group of Ser123(B). Overall, the above results indicate that c-Lys has strong interactions and thus binding with integrin αvβ3.
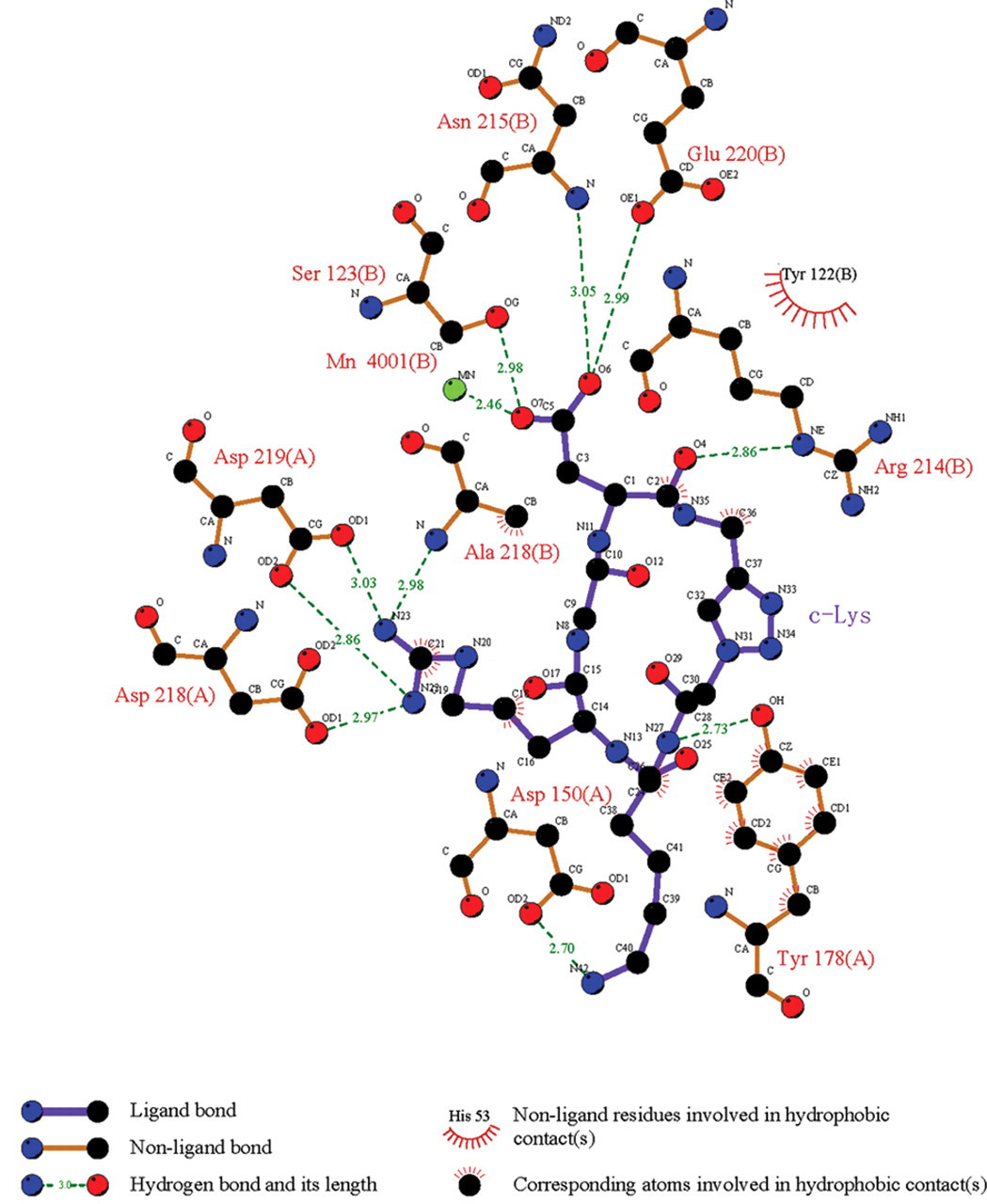
A schematic illustration of the interactions of c-Lys with the residues around the receptor site of integrin αvβ3. Integrin αvβ3 residues and corresponding atoms of c-Lys involved in hydrogen bond and hydrophobic contacts are drawn using the LigPlot program. Dashed lines represent hydrogen bonds, and spiked residues form hydrophobic contacts with the c-Lys inhibitors.
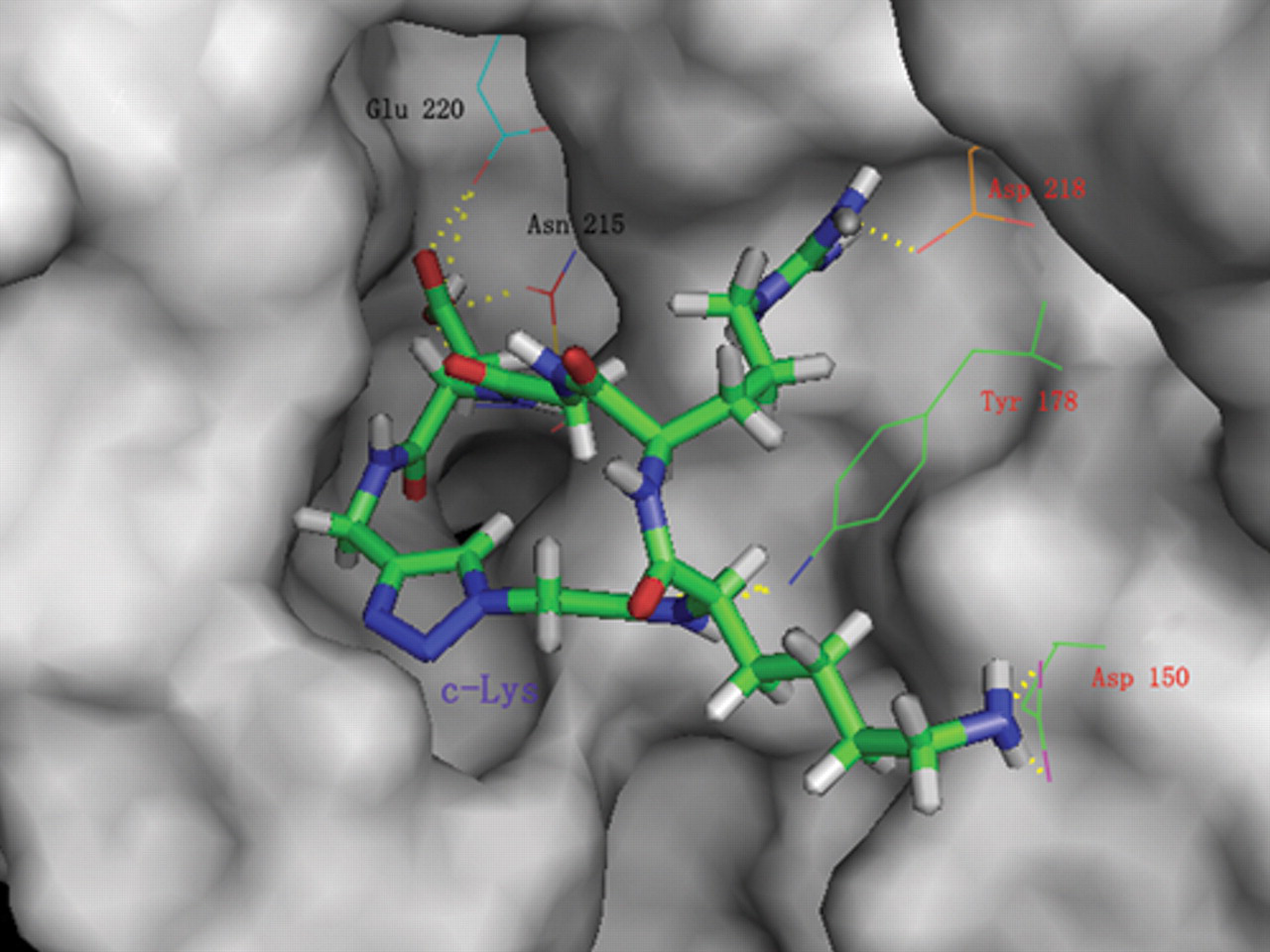
The docking conformation of c-Lys into the active site of the integrin αvβ3 receptor. The ligand, c-Lys, is shown in stick and the important residues around the receptor site surface are shown in line. The αv and β3 residues are labeled red and black, respectively. Hydrogen bond interactions are represented by dashed lines. The c-Lys is accommodated within the binding pocket.
Similar to the case of c-Lys/integrin αvβ3 complex studies, the binding mode of the c-Leu/integrin αvβ3 complex was obtained, which is the most inactive compound in the SPR assay. As shown in Figure 6 , in comparison with the c-Lys/integrin αvβ3 complex, the hydrophobic and hydrogen bond interactions of the c-Leu/integrin αvβ3 complex were less prominent, showing a weak hydrogen bond with only 3 residues—Asp218(A), Arg214(B), and Ser123(B)—of integrin αvβ3. Figure 7 shows that the conformation of c-Leu failed to dock into the integrin αvβ3 receptor site, in which the Asp residue of the RGD motif in c-Leu is pushed further away from the receptor site than it does in c-Lys. Thus, the RGD motif of c-Leu may have a preferred inactive conformation, depending on the effect of its leucyl side chain. As has been mentioned, the molecular docking results have indicated that the c-Lys/integrin αvβ3 complex has a strong multiple hydrogen bond and hydrophobic interactions, whereas the c-Leu/integrin αvβ3 complex only shows weak interactions.
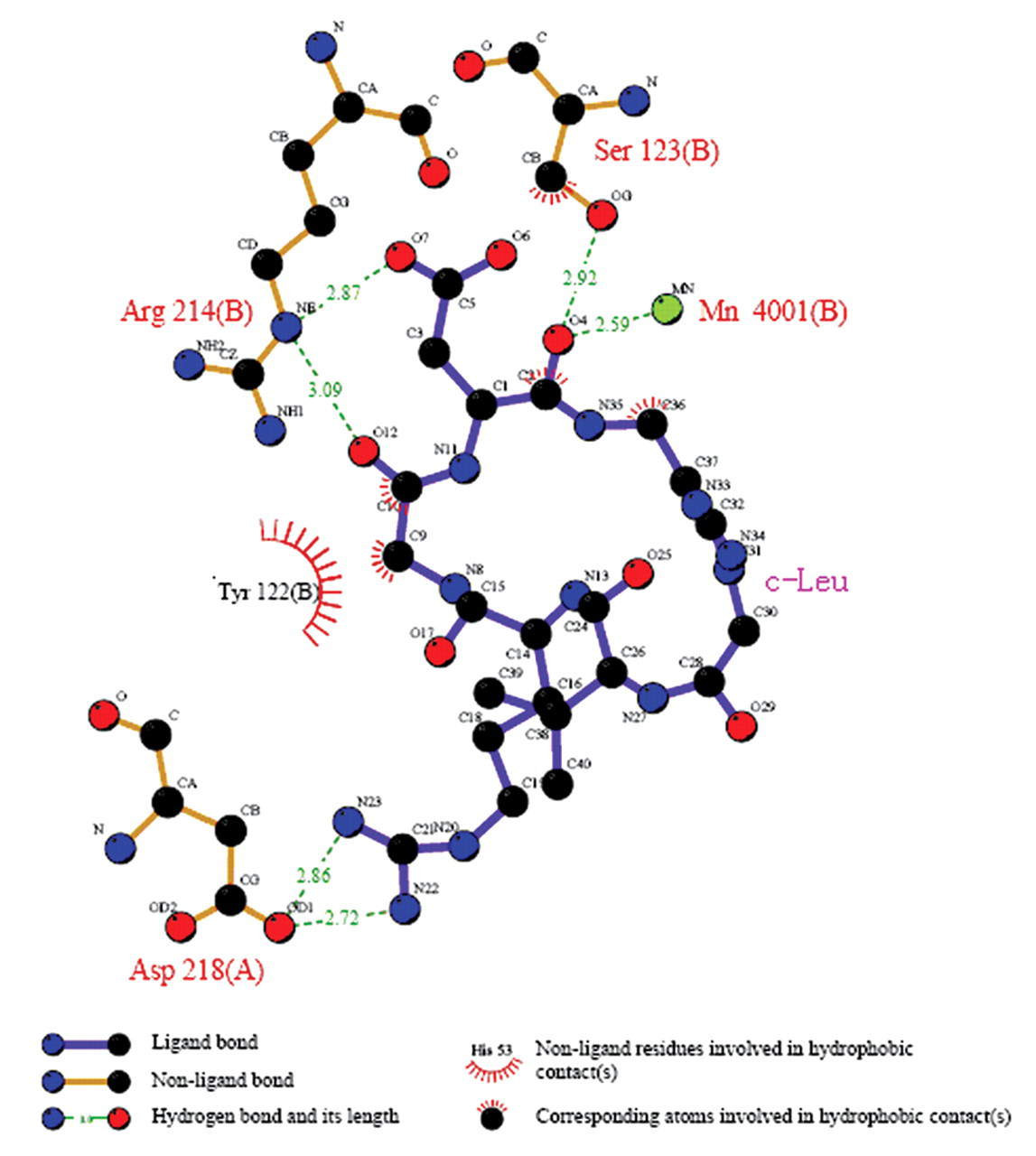
A schematic illustration of the interactions of c-Leu with the residues around the receptor site of integrin αvβ3.
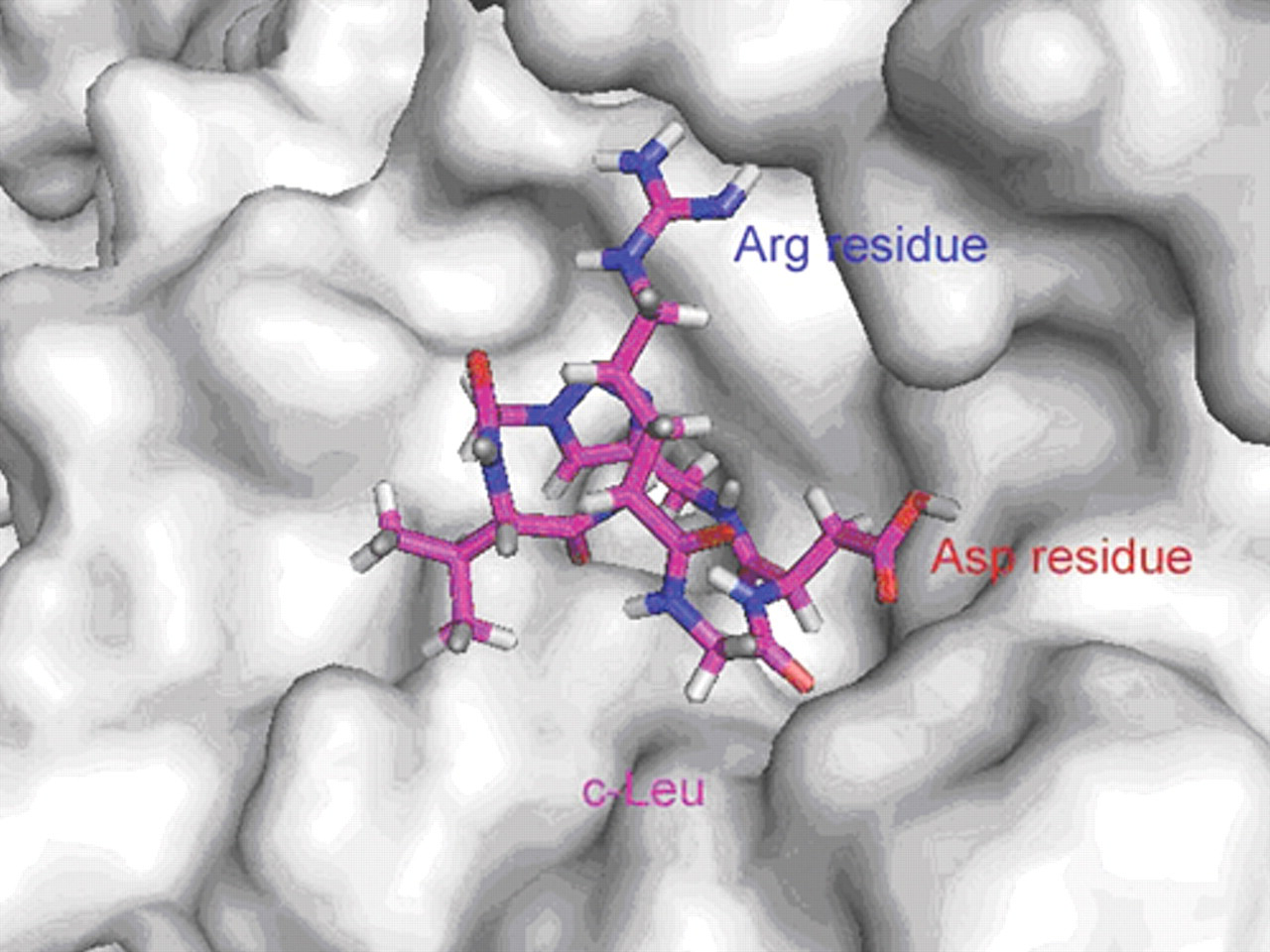
The docking conformation of c-Leu to the active site of integrin αvβ3 receptor. The ligand, c-Leu, failed to bind to the binding pocket domain of the integrin αvβ3, in which the Asp residue of the RGD motif flips out of the binding grooves as a whole. The c-Leu is protruded outside the binding pocket.
Conclusions
In summary, the current work employed SPR technology and molecular docking simulation analyses to study the binding features of several inhibitors to integrin αvβ3. The equilibrium dissociation constant (KD) values evaluated by Biacore X100 for the inhibitor binding to integrin αvβ3 showed that the KD value of cyclopeptide c-Lys is much lower than the reference molecule. In addition, molecular docking technology was performed with the 3D structural model of integrin αvβ3 generated on the basis of the crystal structure of the integrin αvβ3 complex, which has helped us to investigate the inhibitor/integrin αvβ3 binding at the atomic level. The predicted binding sites for the most active cyclopeptide c-Lys were consistent with the reported structure. These results suggested that cyclopeptide c-Lys could be developed as a potential inhibitor for integrin αvβ3. It is hoped that our work will find application in the platform construction for structure-based integrin αvβ3 inhibitor discovery.
Footnotes
Acknowledgements
We are grateful to Prof. Yongzhou Hu for providing the LigandFit package and to Dr. Wenhai Huang and Xiaowu Dong for providing help during the LigandFit docking procedure.