
Abstract
Cardiomyopathies are commonly believed to have genetic origins; however, the connection between cardiomyopathies and cardiovascular diseases remains uncertain. Thus, we employed a Mendelian randomization (MR) approach to investigate the potential causal effects of specific cardiovascular disease subtypes on dilated and hypertrophic cardiomyopathies, focusing primarily on a European population. Summary-level data for cardiomyopathies and other cardiovascular diseases were obtained from public genome-wide association studies. Random-effects inverse-variance weighting was used as the primary analysis, whereas sensitivity analyses, including weighted median, MR–Egger, and multivariable MR methods, were also conducted. A genetic predisposition to atrial fibrillation [odds ratio (OR): 1.33; 95% confidence interval (CI): 1.18–1.50; P < 0.001], heart failure (OR: 3.22; 95% CI: 1.92-5.41; P < 0.001), and hypertension (OR: 1.50; 95% CI: 1.25-1.81; P < 0.001) were causally linked to an increased risk of developing dilated cardiomyopathy. However, there was no direct causal connection between genetically predicted coronary heart disease, pulmonary embolism, or ischemic stroke and the risk of developing dilated cardiomyopathy. In contrast, no significant associations were found between genetically predicted CVD subtypes and the risk of developing hypertrophic cardiomyopathy. Genetically predicted heart failure is significantly associated with the risk of developing dilated cardiomyopathy, underscoring the importance of effective heart failure management for risk prevention. Moreover, individuals with hypertension and atrial fibrillation might have an increased predisposition to dilated cardiomyopathy, highlighting crucial implications for management.
Keywords
Introduction
Cardiomyopathy refers to a diverse group of clinical syndromes that affect myocardial tissue, leading to structural changes in some heart chambers and impaired myocardial wall function, which results in progressive cardiac dysfunction.1,2 Clinical manifestations include heart structural changes, cardiac insufficiency, and arrhythmias. The etiology and subtypes of cardiomyopathy are complex and varied, with hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM) being the most common forms. 3 Cardiomyopathy can affect individuals of all ages.
Recent epidemiological studies and updated evidence have revealed that cardiomyopathy is more prevalent than previously believed. 1 The prevalence of HCM in the general population is estimated to range from 1 in 200 to 1 in 500 people. 4 HCM often occurs in families and typically follows an autosomal dominant inheritance pattern. It is currently thought to be caused by mutations in the genes encoding myocardial contractile proteins. 5 However, these genetic mutations alone are not sufficient to cause the disease phenotype; their effects are influenced by both genetic and nongenetic factors.6,7 DCM constitutes a highly heterogeneous group of diseases resulting from a combination of genetic and nongenetic factors. DCM is the most common type of cardiomyopathy, affecting approximately 1 in 250 people. It is a significant cause of heart failure (HF) with reduced ejection fraction.8,9 Recent research has indicated that DCM is linked to factors such as viral infections, the perinatal period, alcoholism, certain anticancer medications, disruptions in myocardial energy metabolism, and abnormalities in neurohormonal receptors. Nonetheless, the underlying causes and mechanisms remain incompletely understood for a notable proportion of patients. 10
Previously, there was a widespread belief that cardiomyopathy was primarily a genetic disorder, but this belief is not entirely accurate. While DCM and HCM exhibit genetic predispositions, they are not purely genetic diseases;11,12 genetic factors significantly contribute to their development, but they are not the sole determinants. 13 Other cardiovascular conditions, such as hypertension (HTN), coronary artery disease (CAD), HF, and atrial fibrillation (AF), may also influence the onset of cardiomyopathy. However, reliable epidemiological data, particularly across different racial groups, are lacking. To gain deeper insight into the genetic roots of cardiomyopathies and investigate their connection with cardiovascular diseases (CVDs), we conducted an analysis of several genome-wide association studies (GWASs).
Mendelian randomization (MR) involves the use of single-nucleotide polymorphisms (SNPs) as a genetic tool to identify potential causal effects of exposure on diseases, utilizing genetic variation as an instrumental variable. 14 There are many SNP loci in the human genome, and mutations in these loci can lead to numerous diseases. Many phenotypic differences in the human body, susceptibility to drugs or diseases, etc, are associated with SNP loci; thus, we need to screen for SNPs that are strongly correlated with exposure data. 15 Since genetic variation is randomly assigned during meiosis and fertilization and is established before disease onset, it is relatively independent of environmental factors and self-selected behaviors. This minimizes issues with residual confounding and reverse causation. 16 To avoid potential confounding bias, this study utilized the PhenoScanner website to remove the effects of confounders and effectively control for confounders. 17 In addition, we conducted a series of sensitivity analyses such as the leave-one-out method, heterogeneity test, and multiple validity test. These sensitivity analyses help to determine whether the causal conclusions drawn are plausible, while using MR-PRESSO to test the level of multiplicity and removing the weak instrumental variables (F test values >10) to ensure the reliability and accuracy of the results. 18
In this study, tools were derived from large-scale, nonoverlapping GWASs, and summary statistics were obtained from Finnish databases. Multivariable MR analyses were then employed to evaluate the potential causal associations between genetically predicted CVDs and the risk of developing both DCM and HCM. In this study, we utilized tools originating from extensive, nonoverlapping GWASs and summary statistics sourced from FinnGen databases. The aim of this study was to explore whether genetic indicators of specific CVDs are significantly linked to the risk of developing DCM and HCM.
Methods
In MR analyses, genetic variants used as instrumental variables for risk factors must satisfy three core assumptions to yield valid results: (1) the exposure is strongly associated with the genetic variants; (2) the genetic variants are unrelated to confounders affecting the exposure‒outcome association; and (3) genetic variation can affect outcomes exclusively through exposure and not through other pathways. 19 While observational studies are susceptible to confounding factors, MR analysis utilizing genetic instrumental variables can better avoid the influence of confounding factors thereby inferring a causal relationship between exposure and outcome. 20 In conducting MR analyses, it is not possible to demonstrate a causal association between our study's exposures and outcomes simply by using one analytical method; the primary MR analysis is conducted via the IVW method, which assumes that all variables are valid instrumental variables; the method provides the most accurate estimates; and when more than 50% of the weights are derived from valid instrumental variation, the weighted median method can provide consistent estimates. 21 Because IVW is susceptible to potential pleiotropic effects, we used MR-PRESSO as the primary analysis when significant outliers were detected, as MR-PRESSO can clearly identify and correct for potential pleiotropic outliers. 22 For sensitivity analyses, we repeated the analyses via WM and MR Egger regressions. The WM assumes that 50% of the information from the genetic instruments is valid. In contrast, MR Egger regression provides correct estimates, allowing all instrumental variables to be invalid but requiring variants to satisfy the assumption that instrumental variable strengths are independent of direct effects. 23
Data Resources
Summary-level data for DCM (n = 6584) and HCM (n = 2760) were obtained from recent GWAS data. 24 Genetic associations with HTN (n = 412,113), coronary heart disease (CHD) (n = 412,181), and pulmonary embolism (PE) (n = 411,174) were sourced from the FinnGen Consortium. 25 Genetic associations with HF 26 (n = 1,366,492 Europeans, 257,928 East Asians; 26,674 Africans (unspecified), 14,387 Other admixed ancestry), AF 27 (n = 456,348), and ischemic stroke (IS) 28 (n = 1,296,908 Europeans) were extracted from recent GWAS data. Gene effects are inherent in the allele and therefore invariant across genetic and environmental contexts. However, genetic variation varies across populations with different genetic backgrounds, which can lead to spurious associations between genetic variation and outcomes. 29 This phenomenon is particularly common in studies with multiple races or genetic backgrounds. To minimize the influence of potential confounders on the results of the study, the data in this study were mainly obtained from European populations, and the homogeneous population reduced the interference of genotypic and phenotypic data by external factors and improved the accuracy of the study. 20 Homogeneous populations not only help us to better understand the interactions between genes and the environment, which is important for revealing the pathogenesis of diseases but also provide a strong reference for the study of other populations. 30 This MR analysis did not necessitate ethical approval since all analyses were based on publicly available data.
Instrumental Variable Selection
The criteria for selecting instrumental variables were as follows: (1) SNPs were intimately linked to exposure factors and met the genome-wide significance threshold (P < 5 × 108); (2) SNPs showing linkage disequilibrium (r² > 0.001) were removed to maintain the independence of the selected SNPs’ effects; and (3) the F statistic was used to gauge the strength of the association between the instrumental variables and the exposure factors, with a threshold of F > 10 set to effectively mitigate bias arising from weak instrumental variables. 31
Data harmonization ensures that exposure (exposure) and outcome (outcome) SNP effect alleles are concordant, and in GWAS data, SNPs may contain multiple alleles. 16 The harmonization process represents these multiple alleles as double alleles, ensuring their correct treatment in the analysis. Data harmonization also involves the elimination of incompatible SNPs (eg, A/G vs A/C), which are incompatible with exposure and outcome data and can affect the accuracy of the analysis results. Data coordination reduces errors due to inconsistent or incompatible data, thereby improving the accuracy and reliability of Mendelian randomization analyses and providing a solid foundation for further causal inferences. 32
The data were harmonized to ensure that the effects of SNPs on both exposure and outcome corresponded to the same allele. Attempts were made to infer positively stranded alleles via allele frequencies. Efforts have been made to leverage allele frequencies to infer positively stranded alleles and eliminate SNPs with inconsistent alleles. 30 No proxy SNPs were utilized in the current MR analysis.
Statistical Analysis
In our primary analysis, we used random-effects inverse-variance weighting (IVW) to estimate the causal relationships between genetically predicted CVD subtypes and the risk of developing both DCM and HCM. 19 Additionally, we conducted sensitivity analyses via MR‒Egger, weighted median, and simple mode methods. We also applied the MR–PRESSO (MR–Pleiotropy RESidual Sum and Outlier) method to identify potential outliers and recalculate causal estimates after excluding these outliers. 33 We conducted a “leave-one-out” sensitivity analysis to evaluate the influence of each individual SNP on the overall estimate. Owing to differences in different analysis platforms, experimental conditions, enrollment populations, and SNPs, there may be some heterogeneity in the MR analyses, which can bias the estimation of causal effects; therefore to increase the reliability of the findings, the degree of heterogeneity in the individual effects produced by each mutation was assessed via Cochran's Q test. 34 Heterogeneity among the selected SNPs was assessed via the Cochran Q statistic, considering a P value of <0.05 as indicative of significant heterogeneity. If there is heterogeneity, we can use a random-effects IVW model, and if there is no heterogeneity, we can use a fixed-effects IVM model, which does not affect the results of the IVW method despite some heterogeneity. 21 The results are presented as odds ratios (ORs) with corresponding 95% confidence intervals (CIs). 35 Some SNPs, in addition to their association with specific exposure factors influencing outcomes, may also influence outcomes directly or indirectly through other biological pathways, which has important implications for our causal inferences. 36
Ideally, we would like all SNPs to influence the occurrence of the outcome variable through exposure factors only; however, if horizontal pleiotropy exists, those SNPs may influence the occurrence of the outcome variable through influencing exposure factors, which may lead us to overestimate or underestimate the true effect of exposure factors on the outcome variable; therefore, we applied the MR-PLEIOTROPY, MR-PRESSO regression test to monitor potential horizontal pleiotropic effects. 37 We applied MR-RAPS to account for potential weak instrument bias and unbalanced horizontal pleiotropy, which is particularly critical in small-sample scenarios. 38 All the statistical analyses were conducted via the “TwoSampleMR (version 0.4.26)” package in R (R version 4.3.3).
Results
Heart Failure
Genetic predisposition to HF was significantly linked to an increased likelihood of developing DCM (OR: 3.22; 95% CI: 1.92-5.41; P < 0.001; power = 100%) (Figure 1), and the results remained statistically significant after correction for FDR (P < 0.001). Q statistics indicated heterogeneity among the different SNPs, and the MR‒Egger regression intercept for the sensitivity analysis (p value: 0.941) suggested that there was no horizontal pleiotropy. Using MR–PRESSO analysis, we identified three outlier SNPs (rs28579893, rs56094641 and rs55730499) that are associated with cardiomyopathy. Rs56094641, located in the FTO gene, is implicated in cardiomyopathy through its potential impact on metabolic pathways and obesity-related mechanisms, while rs55730499 is linked to atherosclerosis risk via its regulation of the LPA gene. Moreover, rs28579893, located in the HSPB7 gene, has been associated with cardiomyopathies due to its role in protein folding and cellular stress response pathways, which are crucial for maintaining normal heart function. Additionally, MR-RAPS analysis further supported the causal link, showing an increased risk of DCM (OR: 3.37; 95% CI: 2.08-5.45; P < 0.001), with no evidence of significant bias.

The associations between genetically predicted cardiovascular disease (CVD) subtypes and the risk of developing dilated cardiomyopathy (DCM).
A genetic predisposition to HF was inversely associated with the risk of developing HCM (OR: 0.32; 95% CI: 0.12-0.82; P = 0.018; power = 100%) (Figure 2). In the MR–RAPS analysis, the obtained results (OR: 0.30, 95% CI: 0.14-0.68; P = 0.004) furnished evidence supporting a causal association between HF and HCM. However, this association was not significant after FDR correction (p = 0.088). In this study, initial results and MR–RAPS analysis indicated an inverse causal link between the genetic predisposition to HF and the risk of developing HCM. However, after FDR correction, the significance disappeared. Sensitivity analysis indicated no multidirectionality or horizontal pleiotropy. Overall, while the initial findings were promising, the results after FDR correction call for prudent interpretation. More research is needed to confirm the relationship between HF and HCM.
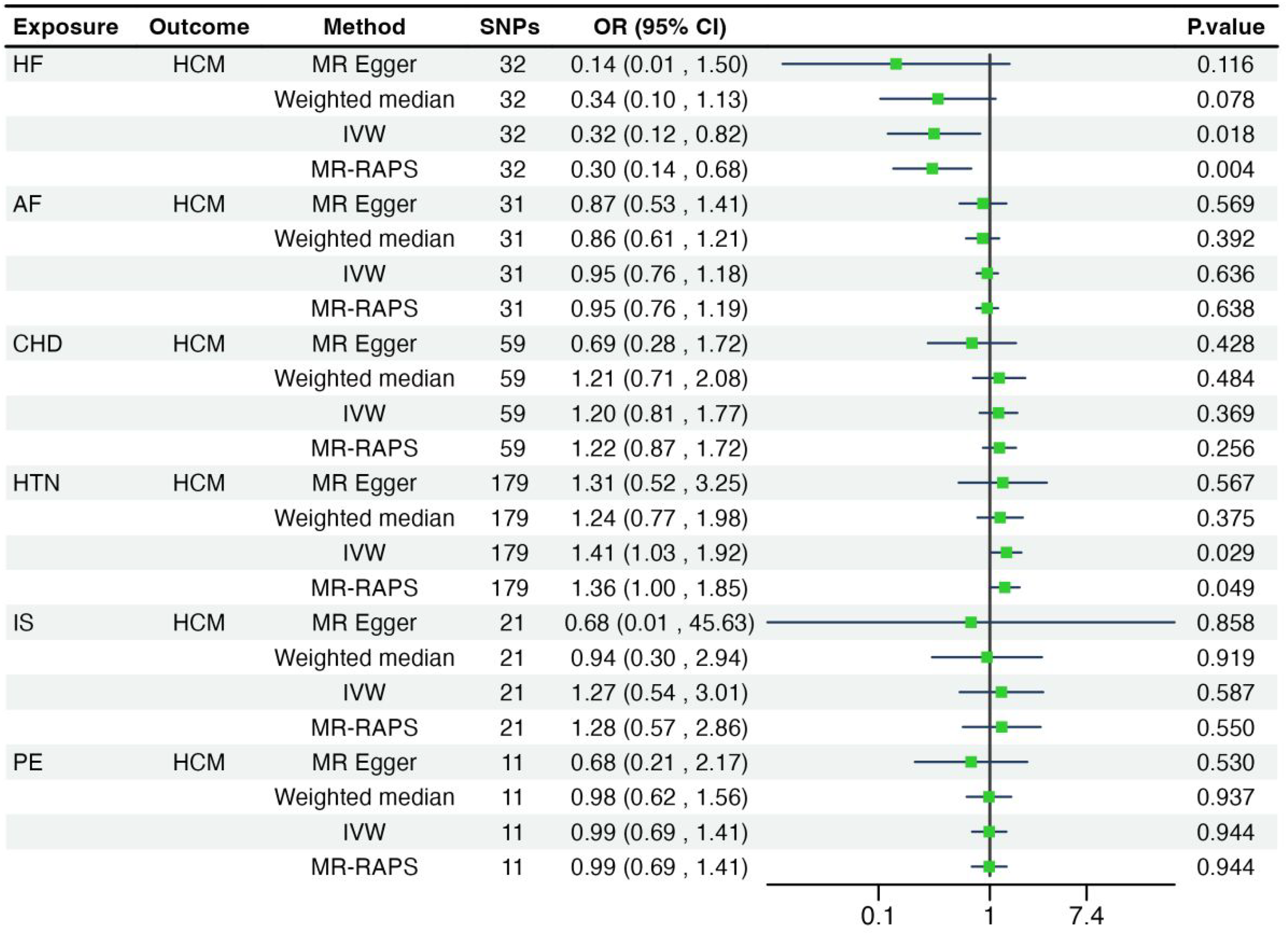
The associations between genetically predicted cardiovascular disease (CVD) subtypes and the risk of developing hypertrophic cardiomyopathy (HCM).
Atrial Fibrillation
There was a positive association between genetically predicted AF and the risk of developing DCM (OR: 1.33; 95% CI: 1.18-1.50; P < 0.001; power = 100%) (Figure 1), and the correlation was statistically significant after correction for FDR (p < 0.001). The Q statistics revealed heterogeneity among the various SNPs, whereas the MR‒Egger regression intercept from the sensitivity analysis (p value: 0.704) indicated no presence of horizontal pleiotropy. In addition, MR–RAPS analysis yielded consistent results, with a positive association between genetically predicted AF and the risk of DCM. The result (OR: 1.33; 95% CI: 1.18-1.51; P < 0.001) further confirmed the causal relationship between AF and the increased risk of DCM.
IVW (OR: 0.95; 95% CI: 0.76-1.18; P = 0.636), weighted median, and MR‒Egger analyses revealed no significant associations between genetically predicted AF and the risk of developing HCM (Figure 2). These results remained non-significant after correction for the false discovery rate (FDR) (p > 0.05). The MR-RAPS analysis also showed no significant association with HCM (OR: 0.95; 95% CI: 0.76-1.19; P = 0.638).
Coronary Heart Disease
IVW (OR: 0.98; 95% CI: 0.77-1.25; P = 0.879), weighted median, and MR‒Egger analyses revealed no substantial associations between a genetic predisposition to CHD and the risk of developing DCM (Figure 1 and Figure 2). The Q statistic revealed heterogeneity among the SNPs, and the MR‒Egger intercept indicated no multidirectionality (p value: 0.193), suggesting the absence of horizontal pleiotropy. The MR-RAPS analysis supported these findings (OR: 0.98, 95% CI: 0.81–1.20; P = 0.876), confirming no significant causal link between CHD and DCM.
No associations were found between genetically predicted CHD (OR: 1.20; 95% CI: 0.81–1.77; P = 0.369) and the risk of developing HCM, with the weighted median method and MR‒Egger producing consistent results. The MR-RAPS analysis further supported these findings (OR: 1.22; 95% CI: 0.87-1.72; P = 0.256). However, none of these results were statistically significant after FDR correction (p > 0.05). These results suggest that genetic predisposition to CHD does not increase the risk of DCM and HCM.
Hypertension
Genetic predisposition to HTN (OR: 1.50; 95% CI: 1.25-1.81; P < 0.001; power = 100%) was positively correlated with the risk of developing DCM, with the weighted median and MR‒Egger methods providing consistent estimates (Figure 1), and the correlation was statistically significant after correction for FDR (p < 0.001). MR-PRESSO did not detect any significant outliers, indicating robust results against potential pleiotropic bias. Furthermore, MR-RAPS provided additional validation (OR: 1.50, 95% CI: 1.26-1.78, P < 0.001). These findings align with the primary results, further supporting evidence of a protective genetic association between HTN and the risk of developing DCM.
Genetic predisposition to HTN was positively associated with the risk of developing HCM (OR: 1.41; 95% CI: 1.03-1.92; P = 0.029; power = 100%) (Figure 2), with no outliers detected and no evidence of heterogeneity or pleiotropy. However, this association was not significant after FDR correction (P = 0.088). MR-RAPS results were consistent (OR: 1.36; 95% CI: 1.00-1.85; P = 0.049). Overall, the initial results indicate a positive link between the genetic predisposition to HTN and the risk of HCM, as supported by MR - RAPS. Nevertheless, after FDR correction, the significance disappeared, suggesting that the initial finding may be false. Therefore, further research involving larger samples or new methods is needed to confirm a causal link.
Ischemic Stroke and Pulmonary Embolism
Genetically predicted IS was not associated with the risk of developing DCM (OR: 0.95; 95% CI: 0.56-1.59; P = 0.836) or HCM (OR: 1.27; 95% CI: 0.54-3.01; P = 0.587) (Figure 1 and Figure 2). There was no significant correlation between genetic susceptibility to PE and the risk of developing DCM (OR: 0.97; 95% CI: 0.80-1.17; P = 0.736) or HCM (OR: 0.99; 95% CI: 0.69-1.41; P = 0.944) (Figure 1 and Figure 2). The MR-RAPS analysis for DCM showed no significant association (OR: 0.97; 95% CI: 0.80-1.17; P = 0.738). Similarly, no significant relationship was found for HCM (OR: 0.99; 95% CI: 0.69-1.41; P = 0.944). The weighted median and MR‒Egger methods yielded consistent estimates, and no significant differences were observed in any of the sensitivity analyses. However, none of the above correlations were statistically significant after correction for FDR (p > 0.05).
Discussion
In this study, we systematically assessed the associations between CVD subtypes and DCM and HCM via MR methods via publicly available large-scale GWAS datasets. Using publicly available large-scale GWAS datasets generated via MR methods, we found that, to some extent, genetic susceptibility to HF, AF, and HTN was associated with the genetic likelihood of developing DCM.
Cardiomyopathies are characterized by genetic and allelic diversity, and often, individual genetic variants, in isolation, are not sufficient to cause the disease phenotype, and their effects are influenced by both genetic and nongenetic factors, which may include common variants and variants with moderate effects. 39 Some patients have a more complex etiology of the disease, requiring the influence of multiple non-Mendelian genetic and nongenetic factors to reach the threshold of the disease, and in addition to significant genetic inheritance, this trend is driven by the aging of the population and the increase in cardiovascular risk factors. Based on morphological and functional features, cardiomyopathies can be categorized into five main types: HCM, DCM, nondilated left ventricular cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, and restrictive cardiomyopathy. 40 The etiology, clinical manifestations, pathophysiology, genetic variants, and types of gene mutations are different for different types of cardiomyopathies.
According to epidemiological data, dilated cardiomyopathy and hypertrophic cardiomyopathy are the most common cardiomyopathies, and etiologic diagnosis and treatment are critical for symptom relief. 41 Therefore, this study focused on analyzing the causal relationships between cardiovascular disease subtypes and dilated cardiomyopathy and hypertrophic cardiomyopathy, which are two common types of cardiomyopathy, to provide evidence-based medicine to optimize the cardiovascular assessment and management of patients with dilated cardiomyopathy and hypertrophic cardiomyopathy.
Cardiomyopathies are complex and diverse disease phenotypes with a wide range of etiologies, pathophysiologies, and clinical processes and are often considered hereditary in nature. 42 This complexity is reflected in the genetic architecture of cardiomyopathies, which include not only those caused by a single disease-causing mutation but also complex traits where susceptibility to environmental triggers is quantitatively influenced by multiple genetic or epigenetic loci with low-exponential effects.5,43,44 Inadequate understanding of the underlying mechanisms and risk factors for the development and progression of cardiomyopathies has hampered the development of effective medical therapies for primary and secondary prevention, and the term “cardiomyopathy,” which is traditionally used to describe only primary diseases of the myocardium, is now also used to describe cardiac dysfunctions associated with concomitant cardiac diseases, including vascular, valvular, inflammatory, and other processes. 45 From a pathological point of view, heart failure begins with myocardial injury that leads to pathological remodeling, resulting in left ventricular enlargement and/or hypertrophy. 46 Initially, compensatory mechanisms dominated by the renin‒angiotensin‒aldosterone system (RAAS), antidiuretic hormone activation, and sympathetic excitation are still able to maintain normal cardiac output through sodium retention, peripheral vasoconstriction, and augmented myocardial contraction, but these neuromuscular mechanisms eventually lead to direct cytotoxicity, causing myocardial fibrosis, arrhythmias, and pump failure. 47
DCM and HCM are two different types of cardiomyopathy, each with different pathophysiologic characteristics. DCM is characterized mainly by enlargement of the cardiac chambers, thinning of the ventricular wall, and fibrous scar formation, and accompanied by attached wall thrombi. 9 Valves and coronary arteries are mostly unchanged, and the histology is a mixture of nonspecific cardiomyocyte hypertrophy, degeneration, and especially different degrees of fibrosis and other lesions. 48 DCM has a slow onset, with no obvious clinical symptoms in the early stage, and at the time of diagnosis, symptoms and signs of congestive heart failure, such as shortness of breath, telangiectasia, edema, can occur in some patients with embolism and sudden death. 49 A wide range of abnormal ECGs are also seen, such as atrial fibrillation, and many abnormal electrocardiograms, such as atrial fibrillation, conduction block and other arrhythmias, are also observed.11,50
However, HCM is characterized mainly by ventricular hypertrophy, the main changes in the myocardium, especially the left ventricular morphology, which is characterized by unequal septal thickening but also by myocardial homogeneous hypertrophy or apical hypertrophy, and the histological characteristics of cardiomyocyte hypertrophy, with different morphologies and disordered arrangements, especially the left ventricular septal part of the change, are obvious. 51 Some patients with HCM have no self-awareness of symptoms, and many have palpitations, chest pain, exertional dyspnea, dizziness and even loss of consciousness. Some patients with HCM may have no conscious symptoms, and many have palpitations, chest pain, and exertional dyspnea. 52 When accompanied by outflow tract obstruction, some patients may experience dizziness or even loss of consciousness due to insufficient diastolic filling of the left ventricle and a decrease in cardiac output. 53
Regarding the relationship between genetically predicted CVD subtypes and the risk of developing cardiomyopathy, numerous studies have consistently shown that cardiomyopathy causes systolic and/or diastolic dysfunction, which impairs the ability of the heart to pump efficiently and ultimately leads to HF.54,55 In our cohort study, we found that HF is the most common clinical manifestation of cardiomyopathy.56,57 Changes in myocardial structure and fibrosis in the heart muscle can disrupt the electrical conduction system, increasing the risk of AF. A global meta-analysis on the prevalence of AF in cardiomyopathy patients estimated that 24% of those with DCM and 19% of those with HCM had AF. 58 In a retrospective real-world study conducted in the United States from 2002 to 2020, data from 634,885 patients with cardiomyopathy revealed that 14,675 cases (2.3%) of DCM and 37,685 cases (5.9%) of HCM were associated with AF status. 59 Patients with cardiomyopathy are susceptible to blood stasis, particularly in the left ventricle and left atrium, due to enlarged chambers and cardiac insufficiency. This stagnant blood flow increases the risk of thrombosis, which can dislodge and cause embolic events such as stroke, PE, and peripheral arterial embolization. 60 Our study demonstrated that genetic susceptibility to heart failure and atrial fibrillation is associated with the genetic likelihood of developing DCM. Hypertension causes myocardial hypertrophy to be mostly symmetrical, and the heart can withstand high blood pressure for a long period, resulting in a gradual increase in the left ventricle load. The left ventricle, owing to compensation and gradual hypertrophy and dilatation of the ventricular wall, often results in more homogeneous thickening of the whole ventricular wall. 61 Thickening of the myocardial echogenicity does not lead to obvious changes, and generally does not cause left ventricular outflow tract obstruction. 62 In HCM, left ventricular hypertrophy is more pronounced, mostly asymmetric, with marked septal hypertrophy, possible obstruction, and increased myocardial echogenic intensity at the lesion site, which is grossly glassy or coarse speckled. 60 In our study, we also confirmed that genetic susceptibility to hypertension was not associated with genetic susceptibility to HCM but was associated with genetic susceptibility to DCM. Coronary artery stenosis or occlusion results in myocardial ischemia, and repeated episodes of ischemia cause myocardial cell death and fibrosis, leading to thinning of the ventricular walls and enlargement of the heart chambers. 63 Patients with CHD may have reduced myocardial motion at the ischemic site, which manifests as segmental decreased motion, whereas dilated cardiomyopathy is characterized by generalized diffuse myocardial decreased motion, thinning of the ventricular septum, and reduced motion amplitude of the left ventricular posterior wall.8,64 Additionally, in our study, there was no association between genetic predisposition to and genetic predisposition to cardiomyopathy.
Using MR analysis, our findings offer genetic evidence underscoring the necessity for increased support and attention not only in terms of prognosis and treatment but also in the prevention of cardiomyopathy within this population. However, our study has several limitations. When genetic variation is weakly associated with exposure, this can lead to biased estimates from MR models, called weak instrumental variable bias. 65 In MR models, the F statistic is often used to evaluate the strength of instrumental variables. To ensure that the instrumental variables in the model have sufficient strength, existing MR models usually use the genome-wide significance level (P < 5 × 10-8) as a threshold to screen the instrumental variables to avoid potential weak instrumental variable bias. 64 In addition, when the average strength of instrumental variables in MR models is weak, some improved MR models, such as IVW based on modified weights and Egger's test, can be applied to correct for potential weak instrumental variable bias. 66
In practical research, we can identify polyvalent instrumental variables via visualization methods such as scatter plots and funnel plots combined with statistical tests. Commonly used tests include the Q-statistic test, MR‒PRESSO test and heterogeneity test. After the polytomous instrumental variables are identified and eliminated, the Q statistic test, PRESSO test, MR‒Egger intercept term test, etc, can be used to evaluate the polytomousness of the remaining instrumental variables. 67 If there is no heterogeneity in the proportion estimates corresponding to the remaining instrumental variables, it suggests that there are no multinomial instrumental variables, and then the MR model constructed based on the assumption that no multinomial instrumental variables can be applied to estimate the effects. 32 The causal effects can also be estimated directly via MR models to correct for polytomous bias, and methods based on pooled data include MR‒Egger and median-based estimation. 21
The exposure variance explained by genetic variance is only a part of the total exposure variance, so genetic variance is used as an instrumental variable for exposure to estimate its causal effect on the outcome. 68 The effect value obtained is only the effect of this part of the exposure variance on the outcome determined by the instrumental variance. MR modeling can provide statistical clues as to whether there is a causal association between the exposure and the outcome and provide a theoretical basis for the subsequent more definitive experimental. 69 The MR model can provide statistical clues to the causal association between exposure and outcome and provide a theoretical basis for subsequent more definitive experimental studies and mechanism exploration, but no single research method can completely clarify the causal relationship. 70 However, no single research method can completely clarify the causal relationship, and the real causal relationship should be explored by combining the biological mechanism of the disease, the results of comprehensive experimental and clinical studies, and other evidence. 68
Although our genetic data were sourced from large GWASs with diverse samples, enhancing statistical power to detect small effects in complex phenotypes, our findings exhibited partial heterogeneity. In MR analyses, results from other methods (MR Egger, weighted median, simple mode, and weighted mode) were not entirely consistent with those from the IVW method. This heterogeneity may arise from potential nonlinear relationships or stratification effects due to variations in health status, age, or sex. However, the majority of estimates align with the sensitivity analysis, supporting the reliability of our findings and suggesting they are not due to chance. We also considered the challenges posed by small sample sizes, particularly in the HCM and DCM datasets, which are prone to statistical instability and type II errors. To address this, we applied MR-RAPS analysis, F-statistics, and leave-one-out sensitivity analyses to enhance statistical robustness. Future research will focus on optimizing these methods to minimize bias and uncertainty associated with small sample sizes. While this study provides robust evidence for the causal relationships between CVD subtypes and cardiomyopathy, its findings are primarily applicable to European populations. Genetic, environmental, and healthcare differences across populations may limit the generalizability of these results. Future research should replicate these analyses in diverse populations to evaluate the broader applicability of these findings.
Conclusion
This MR study demonstrated that genetically predicted genetic susceptibility to HF, AF, and HTN was associated with the genetic likelihood of developing DCM, whereas genetic susceptibility to these CVD subtypes was not associated with HCM. In addition to the known etiologic factors, the impact of the CVD subtypes included in this study on the risk of developing DCM reflects their potential usefulness for the development of therapeutic strategies and warrants further investigation in future larger MR analyses.
Footnotes
Acknowledgments
We want to acknowledge the participants and investigators of the FinnGen study. We are grateful to all the GWAS Consortium authors and participants who contributed the summary statistics. The design and implementation of this study, all the research analyses, the drafting and editing of the paper, and the final content are the sole responsibility of the authors.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability
All GWAS and FinnGen summary statistics analyzed in this study are publicly accessible. Genomic data for HF (GCST90162626), AF (GCST90043977), IS (GCST90104540), DCM (GCST90296097), and HCM (GCST90296096) can be accessed through the GWAS Catalog (https://www.ebi.ac.uk/gwas/downloads/summary-statistics/). Data for CHD (https://storage.googleapis.com/finngen-public-data-r10/summary_stats/finngen_R10_I9_CHD.gz), HTN (https://storage.googleapis.com/finngen-public-data-r10/summary_stats/finngen_R10_I9_HYPTENS.gz), and PE (https://storage.googleapis.com/finngen-public-data-r10/summary_stats/finngen_R10_I9_PULMEMB.gz) were obtained from FinnGen (![]() ).
).