
Abstract
Inherited thrombocytopenia is a group of hereditary diseases with a reduction in platelet count as the main clinical manifestation. Clinically, there is an urgent need for a convenient and rapid diagnosis method. We introduced a high-throughput, next-generation sequencing (NGS) platform into the routine diagnosis of patients with unexplained thrombocytopenia and analyzed the gene sequencing results to evaluate the value of NGS technology in the screening and diagnosis of inherited thrombocytopenia. From a cohort of 112 patients with thrombocytopenia, we screened 43 patients with hereditary features. For the blood samples of these 43 patients, a gene sequencing platform for hemorrhagic and thrombotic diseases comprising 89 genes was used to perform gene detection using NGS technology. When we combined the screening results with clinical features and other findings, 15 (34.9%) of 43patients were diagnosed with inherited thrombocytopenia. In addition, 19 pathogenic variants, including 8 previously unreported variants, were identified in these patients. Through the use of this detection platform, we expect to establish a more effective diagnostic approach to such disorders.
Introduction
Inherited thrombocytopenia comprises a group of rare hereditary diseases characterized by a reduction in the platelet count. 1,2 Most subtypes of inherited thrombocytopenia have a very low incidence. The main symptom reported is mucocutaneous bleeding or abnormal hemostasis after trauma. Female patients have excessive menstrual bleeding, and some patients have comorbid developmental malformations of bone and heart. 2 –4 The majority of patients display various bleeding tendencies, and some patients do not have obvious bleeding symptoms but still exhibit a moderate reduced platelet count. However, specific detection methods for inherited thrombocytopenia are still lacking in the clinic. Usually, first-line hospitals with higher diagnostic levels and facilities can integrate morphological and functional tests to perform preliminary screening of this group of patients and discover possible diagnostic clues. 5 –7 Next, traditional sequencing technology is often performed to identify possible pathogenic genes in these patients. 6,7 However, the entire process requires a longer time and increases the burden on patients and the public health system. Furthermore, even using these diagnostic methods, a large percentage of patients with inherited thrombocytopenia still cannot obtain definitive diagnoses. These patients are usually misdiagnosed with immune thrombocytopenia (ITP) and receive inappropriate treatment.
The widespread application of next-generation sequencing (NGS) technology has highlighted its advantages of high efficiency and high throughput. 8 –11 Establishing a partnership among a central hospital and multiple community hospitals can reduce the costs for each instance of NGS; therefore, more patients can undertake the sequencing expense. 12 –14 Based on NGS, we established a high-throughput gene sequencing platform for the diagnosis of bleeding and thrombotic diseases. The platform covers 65 types of bleeding and thrombotic diseases, including 18 types of known inherited thrombocytopenia. A total of 24 genes have been reported to be associated with these 18 diseases in the platform. We performed gene analysis with this sequencing platform for 43 patients with suspected inherited thrombocytopenia who were treated at 1 of 5 hospitals in Suzhou District, China. The purpose of this study is to identify a proper screening method and to establish new diagnostic procedures with the goal of increasing the early diagnosis rate for patients with inherited thrombocytopenia.
Materials and Methods
Patient
Forty-three unrelated patients with suspected inherited thrombocytopenia were enrolled. The clinical data of the patients were evaluated by a team of professional hematologists to exclude individuals with immune or other acquired thrombocytopenia. The patient cohort included 26 men and 17 women. The age of patients ranged from 2 to 55 years old, with a median age of 38. Investigations abided by the Declaration of Helsinki and were approved by the Ethical Committee of the First Affiliated Hospital of Soochow University, Suzhou, China. All patients signed informed consent.
Construction of the NGS Platform
Based on work described by Simeoni, 14 we designed a gene sequencing platform for hemostasis and thrombosis (HAT platform) that covered 65 known hereditary bleeding/thrombotic diseases and included 89 related genes. Among them, 78 genes were adopted directly from the Simeoni scheme, and the other 11 genes were supplemented according to the latest literature reports. 15,16 This platform included 24 genes related to 18 types of inherited thrombocytopenia (Table 1). The Ion AmpliSeq Designer primer design tool (Thermo Fisher Scientific, Waltham, Massachusetts) was used to design multiplex polymerase chain reaction (PCR) primers for these target genes.
Genes Included in the NGS Platform for Molecular Screening of Inherited Thrombocytopenia.
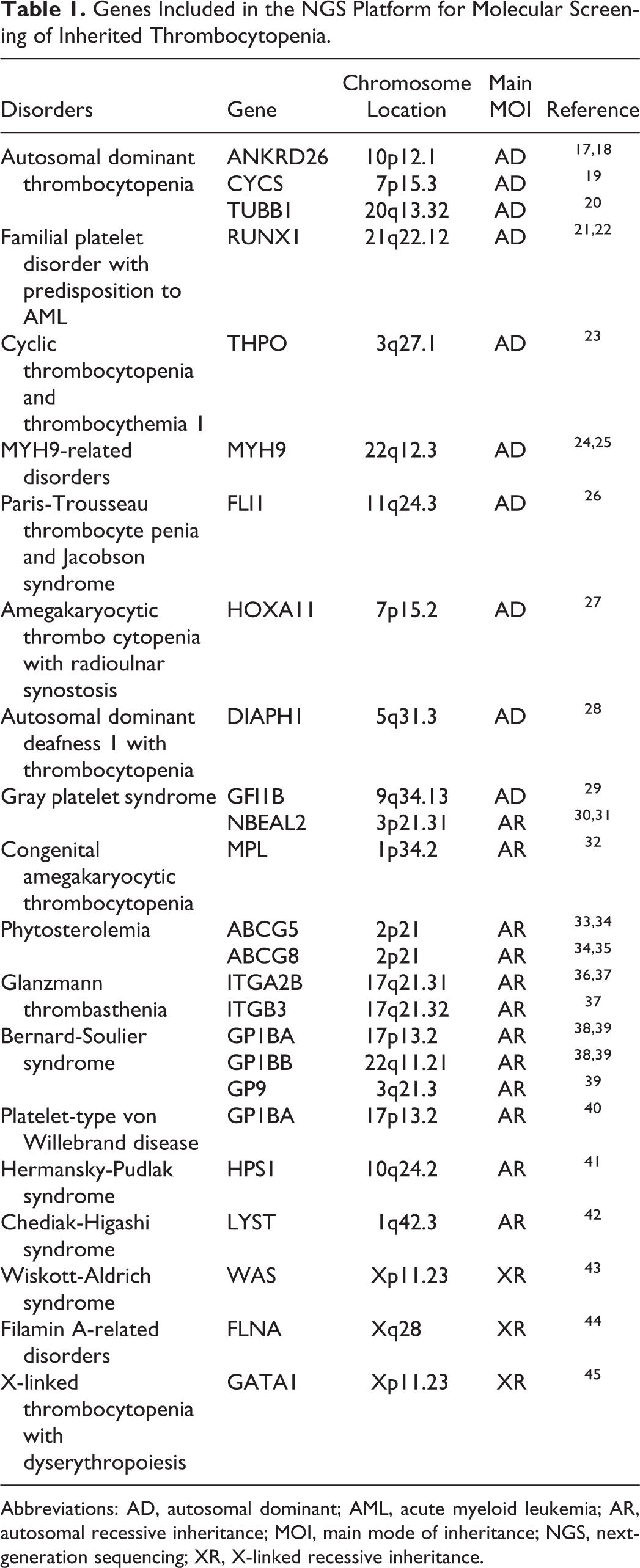
Abbreviations: AD, autosomal dominant; AML, acute myeloid leukemia; AR, autosomal recessive inheritance; MOI, main mode of inheritance; NGS, next-generation sequencing; XR, X-linked recessive inheritance.
Sample Preparation and Sequencing
Peripheral blood was collected from patients into EDTA-anticoagulant tubes. Sample DNA was extracted using a whole blood DNA reagent kit (QIAGEN, Hilden, Germany). The DNA concentration in the samples was measured using a Qubit dsDNA reagent kit (QIAGEN). Then, sample DNA was diluted to 5 ng/µL and amplified using HotStart ReadyMix (QIAGEN). The PCR products were normalized using a SequalPrep Normalization kit (Invitrogen, Carlsbad, California). Enzymatic digestion of the PCR products was performed using an Ion Shear Kit (Thermo Fisher Scientific, Waltham, Massachusetts), DNA ligation was performed using an Ion Xpress Plus Fragment Library Kit (Thermo Fisher Scientific), and DNA labeling was performed using an Ion Xpress Barcode Adapters 1-16 Kit (Thermo Fisher Scientific). DNA fragments approximately 200 bp in size were screened using E-Gel, and DNA library quantitation was performed using an ion library.
Quantitation Kit (Invitrogen)
Next, emulsion PCR (em-PCR) was performed using the Ion Template preparation and One Touch kits (Thermo Fisher Scientific). Positive templates were enriched using MyOne C1 beads (Thermo Fisher Scientific, Waltham, Massachusetts). The above 2 steps were both performed in an Ion OneTouch ES machine (Thermo Fisher Scientific). The sequencing reaction was performed using Ion Torrent 540 chip in an Ion S5 sequencing machine (Thermo Fisher Scientific).
Quality Control of HAT Platform
To examine the validity and accuracy of the HAT platform, we evaluated 6 samples from patients with known gene defects; the 6 cases included MYH9-related disorders (MYH9-RD; n = 1), Glanzmann thrombasthenia (n = 1), Bernard-Soulier syndrome (BSS; n = 1), phytosterolemia (n = 1), Gray platelet syndrome (n = 1), and von Willebrand disease (VWD; n = 1). Among these 6 cases, 2 are homozygous missense variants, 1 is a heterozygous missense variant, 1 is a heterozygous nonsense variant, 1 is a compound heterozygous missense variant, and 1 is a heterozygous frameshift variant. The gene variants of these patients were all detected by Sanger sequencing technology, and the gene variants conformed to their clinical features. Using the HAT platform, the gene variant detection rate of these 6 patients was found to be 100%, and the accuracy rate was 100%. Theoretically, the Ion Torrent 540 chip used for sequencing could carry 20 samples each time. However, in the actual application, 8 to 12 samples were sequenced on each chip to ensure higher sequencing depth. In addition, 1 case among the abovementioned 6 positive controls was randomly added and used as an internal control for the sequencing.
Screening and Analysis of Pathogenic Variants
The sequencing data were preliminarily analyzed using Ion Torrent Server software and were mapped to the Reference Human Genome hg19. The whole data volume, mean sequencing depth, and reference sequence matching degree were obtained. After preliminary gene variation data were obtained, screening for pathogenic genes was performed using the following method. First, variations that caused amino acid changes (exon variants), including nonsense variants, missense variants, and small fragment insertions/deletions, were considered; conversely, synonymous variants and intron variants were ignored (in some cases, variants in splicing regions might cause diseases; therefore, splicing region variations were included in the next screening step). As inherited thrombocytopenia are rare diseases with a very low incidence, variants with minor allelic frequencies (MAFs) > 0.02 were excluded. 16 Among variants with MAFs < 0.02, PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) 46 and SIFT (http://sift.bii.a-star.edu.sg/) 47 were used to predict the functional effects and pathogenicity of variants on amino acid changes. 15,48,49 Several human gene databases were referenced, including HGMD (Human Genetic Mutation Database), COSMIC (Catalogue of Somatic Mutations in Cancer), OMIM (Online Mendelian Inheritance in Man), ClinVar, Pathogenic Variant Assessor, Asian population genome, and Chinese 1000 Genomes Project. According to the guidelines for the classification of hereditary pathogenic genes by the American College of Medical Genetics and Genomics, 50 gene variants detected were classified as “pathogenic,” “likely pathogenic,” “benign,” “likely benign,” or “variant of uncertain significance.” The pathogenic variants detected were verified by Sanger sequencing. Novel pathogenic variants were further confirmed by a literature survey. For patients with pathogenic variants, their parents’ blood samples were also collected and sequenced by the Sanger method to determine whether the variants originated in the germ line. 48,49
Results
Patient Features
Of the 43 patients, 25 had various bleeding symptoms, and the remaining 18 patients only exhibited low platelet counts and did not present obvious bleeding. Among the patients with bleeding symptoms, 64% (16/25) had mild levels of bleeding. Regarding the degree of reduced platelet count, 41.9% (18/43) of patients had a moderately low platelet count (20 × 109/L < PLT < 50 × 109/L), and 46.5% (20/43) of the patients had a mildly low PLT count > 50 × 109/L. The clinical history of the patients was evaluated according to the assessment table of the hereditary features (Table 2), which is based on Drachman review. 51 According to this scoring table, a score ≥3 means that in addition to an extensive history of bleeding, there is at least 1 clinical feature that may point to hereditary thrombocytopenia. A score ≥6 points means that the patient has multiple clinical manifestations that are different from common ITP. In contrast, genetic defects are more likely to cause the disease in these patients. In this study, 95.3% (41/43) of the patients had a score ≥3, and 37.2% (16/43) had a score ≥6.
Assessment Table of the Hereditary Features of Patients With Thrombocytopenia.

Abbreviation: ITP, immune thrombocytopenia.
Among the 43 patients, 27 (62.8%) were previously diagnosed with ITP, and 25 patients had undergone immunosuppressive therapy, such as dexamethasone or cyclosporine. Three patients (case 3, 4, 11, see Table 3) underwent gene detection by the Sanger sequencing previously, but no pathogenic variants were detected.
Clinical Features of the 43 Patients in the Cohort With Suspected Inherited Thrombocytopenia.
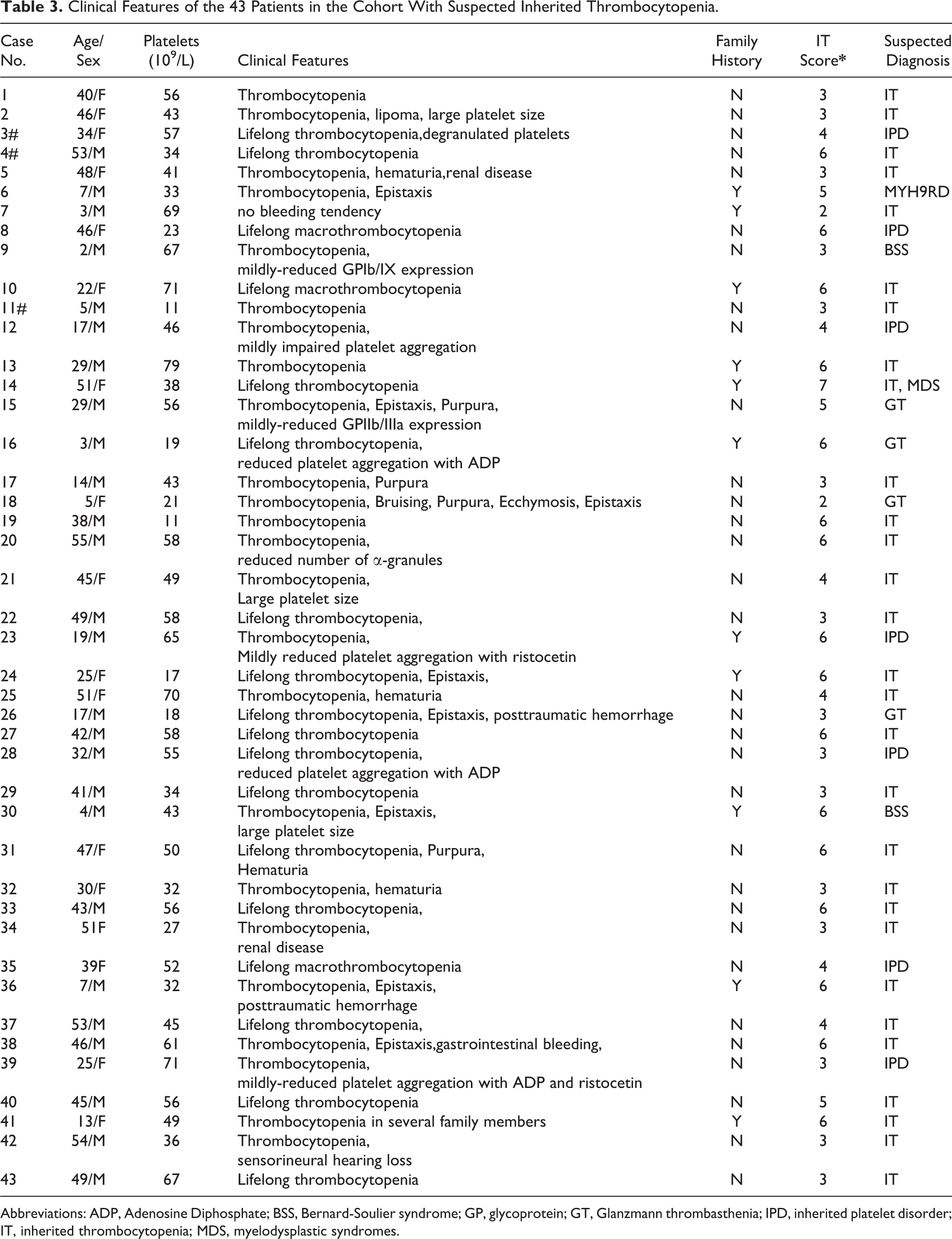
Abbreviations: ADP, Adenosine Diphosphate; BSS, Bernard-Soulier syndrome; GP, glycoprotein; GT, Glanzmann thrombasthenia; IPD, inherited platelet disorder; IT, inherited thrombocytopenia; MDS, myelodysplastic syndromes.
Analysis of NGS Quality
A total of 2064 pairs of primers were designed using Ion AmpliSeq Designer and synthesized; these primers covered 99.1% of the exon regions, splicing regions, and intron regions within approximately 20 bp of both ends of the exons in the related genes. Figure. 1 shows the coverage degree and sequencing depth in an actual assay that uses the Ion Torrent 540 chip. The effective region in the sequencing chip reached 93% of which 95% was connected to the library. The final valid library was 73%, with a total of 99 768 769 read fragments, and the mean length of the fragments was 185 bp. The human genome (hg19) was used as the reference sequence for the abovementioned gene sequences. A total of 28.6 Mb of fragments matched the reference sequence, which accounted for 96.3% of the total number of sequences. The average sequencing depth was 1253×; fragments with more than 100× accounted for 97.6%; and fragments with more than 500× accounted for 92.7%. The detection rate of gene variants of the positive internal control was 100%. The consistency of the detection results between Sanger sequencing and NGS was 99.1%.
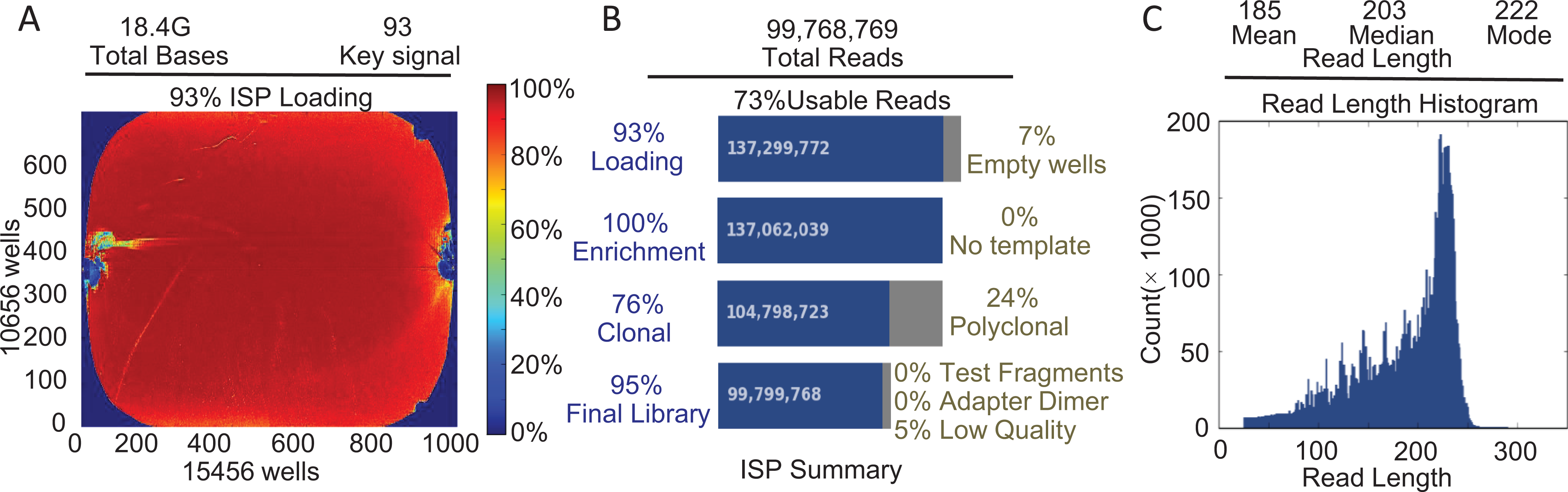
Sequencing output of the HAT NGS platform.
General Performance of the HAT Platform
Overall, 102 gene variants were detected in 43 samples, which were distributed across 35 genes. A total of 99 (97.1%) variants were located in exon regions and 3 (2.9%) were sites in splicing regions. Among the variants in the exon regions, there were 21 (20.0%) homozygous missense, 43 (43.4%) heterozygous missense, 2 (2.0%) homozygous nonsense, 5 (5.0%) heterozygous nonsense, 3 (3.0%) homozygous frameshift, and 15 (15.2%) heterozygous frameshift variants.
Combining the NGS results with the patients’ clinical characteristics and other test results, we identified 15 (34.9%) patients who carried pathogenic variants related to inherited thrombocytopenia. Sixteen (37.2%) had potentially pathogenic variants, and the remaining 12 (27.9%) had benign or uncertain genetic variants.
The 15 patients with inherited thrombocytopenia pathogenic variants were included in 9 disease types (Table 4) and involved 19 genetic variants, 5 of which were described for the first time. Regarding the genetic characteristics, 8 of 15 cases were autosomal recessive (4 cases had homozygous variants and 4 cases had compound heterozygous variants), while the other 7 cases were autosomal dominant (see Table 4 for details). Blood samples were also collected from the parents of 9 patients, and Sanger sequencing was performed to confirm the sources of the disease-causing variants. We failed to obtain parental blood samples from the remaining 6 cases for various reasons. Fingernail samples were collected from all 15 patients, and pathogenic variants were validated using Sanger sequencing. The consistency rate of the results between the fingernail and blood samples was 100%, which confirmed the germline sources of the pathogenic variants in these patients.
Clinical Features and Pathogenic Variants Identified in 15 Patients With Inherited Thrombocytopenia.
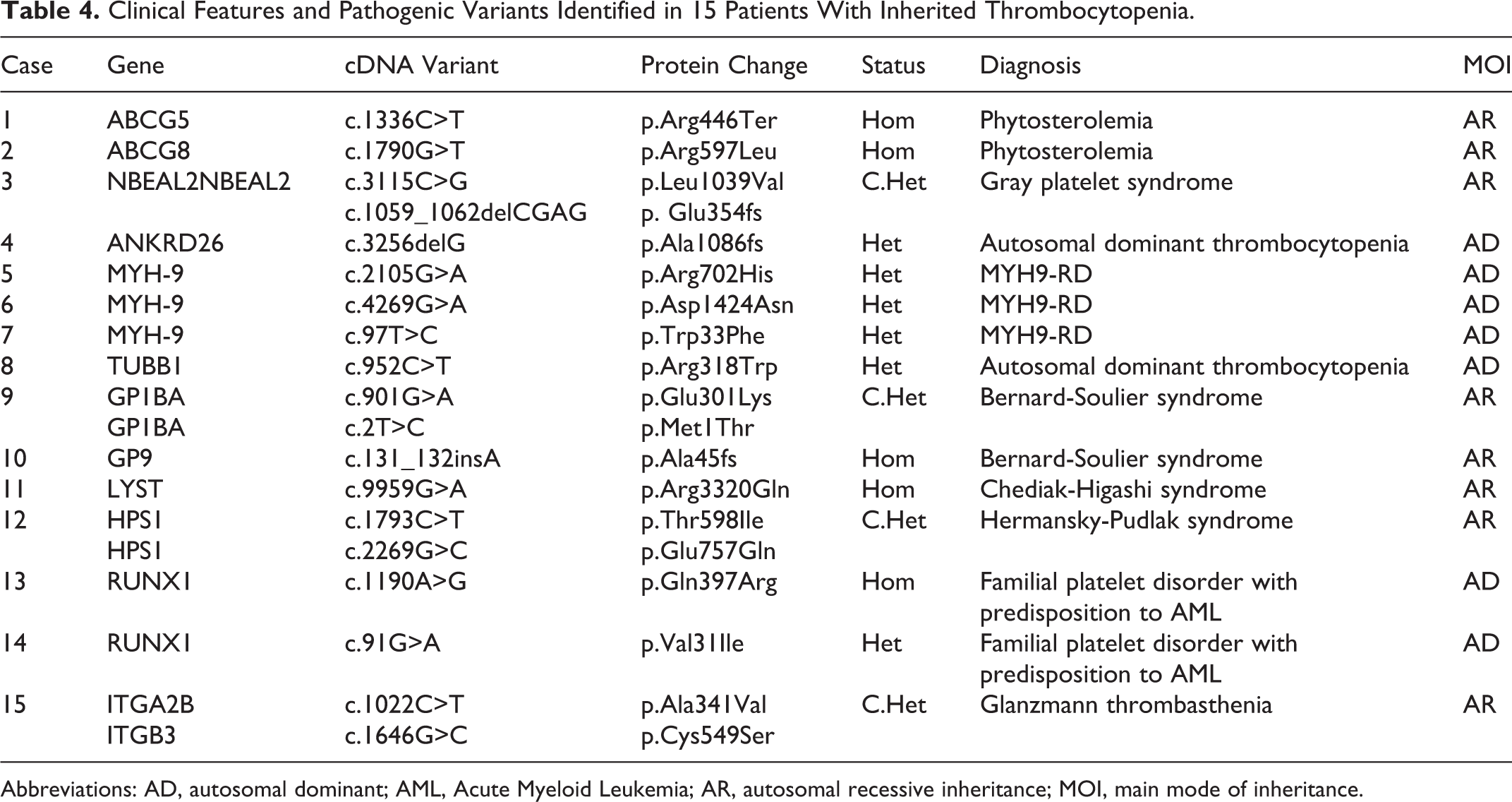
Abbreviations: AD, autosomal dominant; AML, Acute Myeloid Leukemia; AR, autosomal recessive inheritance; MOI, main mode of inheritance.
Table 3 shows 19 pathogenic variants detected in samples from 15 patients. The variants previously unreported include c.2T>C (p.Met1Thr) on GP1B, c.1022C>T (p.Ala341Val) on ITGA2B, c.1646G>C (p.Cys549Ser) on ITGB3, c.131_132insA (p.Ala45 fs) on GP9, c.97T>C (p.Trp33Phe) on MYH9, c.1790G>T (p.Arg597Leu) on ABCG8, c.1793C>T (p.Thr598Ile) on HSP1, and c.3256delG (p.Ala1086 fs) on ANKRD26. The other variants include missense on ABCG5, 1 deletion on NBEAL2, 1 missense on LYST, 1 missense on HSP1, 2 missenses on RUNX1, 1 missense on GP1B, and 2 missenses on MYH-9, the final 2 of which have been reported as hotspot variations that cause nonmuscle myosin heavy chain 9-related disease (MYH9-RD). 52 Additionally, 28 of the 43 cases did not possess definitive pathogenic variants, and we listed the clinical characteristics and gene results of these 28 patients in Table 3.
Discussion
Inherited thrombocytopenia consists of a group of rare diseases that involve dozens of related genes with inheritance patterns such as autosomal dominant, autosomal recessive, and X-linked recessive inheritance. 53 Clinically, these diseases can manifest as skin/mucous bleeding, menorrhagia, and other symptoms. Some patients may have features such as a reduction/absence of bone marrow megakaryocytes and morphology changes of platelets in peripheral blood smears, which often rely on highly experienced hematology pathologists to assess. Furthermore, there is still demand for evidence of molecular pathology to reach a definitive diagnosis. 15,49
The clinical features of the majority of patients with inherited thrombocytopenia can be mildly to moderately low platelet count and various bleeding manifestations. Due to the lack of specific clinical symptoms other than low platelet count and the lack of convenient diagnostic methods, patients with inherited thrombocytopenia are often misdiagnosed with ITP and receive unnecessary immunosuppressive therapy that could be not only ineffective but also harmful to these patients. Among the 15 patients carrying pathogenic gene variants discovered in this study, 11 had been misdiagnosed with ITP at several hospitals. The oldest patient had been misdiagnosed with “refractory ITP” for 16 years and underwent ineffective treatments that unfortunately increased the patient’s pain and financial burden.
The general gene detection procedure involves the selection of a candidate gene through morphological and functional examinations. Next, Sanger sequencing is performed to discover the variant (if any) in the specific gene. 26 For patients with inherited thrombocytopenia, this procedure does not seem to be effective. First, because the suspected diagnosis may be incorrect and result in the selection of the wrong gene to be sequenced, it is obviously impossible to detect the pathogenic variants. This happened in case 4 and 11 in the cohort. In addition, due to technical limitations, Sanger sequencing is sometimes difficult to detect heterozygous small fragment deletions. Case 3 in our study is similar to this situation.
According to our experience, in the case of patients with suspected hereditary thrombocytopenia, physicians should first assess whether patients have characteristics of hereditary defects. If hereditary features are present, screening for gene defects using NGS technology in conjunction with morphological and functional examinations will be more efficient than the traditional diagnostic procedure. We developed an assessment table for evaluating the hereditary features of patients with bleeding or low platelet count based on Drachman review. 51 According to this table, the patient’s family history and bleeding history during childhood is the most important hereditary feature. The efficacies of immunotherapy and platelet transfusion therapy are also useful for distinguishing ITP from inherited thrombocytopenia. For actual application, it should be considered that the reduced platelet count in patients with scores ≥3 probably results from gene defects, and these patients should be persuaded to undergo genetic testing. Patients with scores ≥6 are highly suspected to have inherited thrombocytopenia. These patients are strongly recommended to undergo NGS while receiving other examinations, such as platelet morphological and functional examinations, to confirm their diagnosis.
Among the 43 patients who underwent NGS analysis, 15 finally obtained a clear molecular pathological diagnosis: MYH9-RD (n = 3), phytosterolemia (n = 2), familial platelet disorder with predisposition to acute myeloid leukemia (n = 2), BSS (n = 2), variant-form of GT (n = 1), platelet-type VWD (n = 1), Chediak-Higashi syndrome (n = 1), Hermansky-Pudlak syndrome (n = 1), and autosomal dominant thrombocytopenia (n = 2). In these 15 patients, there were 19 gene variants detected, 8 of which were newly described.
Cases 1 and 2 had gene variants in ABCG5 and ABCG8, respectively. The missense variant c.1790G>T (p.Arg597Leu) in ABCG8 was reported for the first time. The patient (case 2) was a 46-year-old female who had a long-term (>5 years) mild reduction in the platelet count but did not exhibit other symptoms. She had yellow lipoma on the face for the past 6 months, and the most recent blood smear examination showed some giant platelets. Peripheral blood assessment using high-performance liquid chromatography showed that the phytosterol level of the patient was significantly higher than the levels in healthy individuals. The symptoms above conformed to phytosterolemia. 54,55 The pathogenic variant was also present in both parents who were carriers of this variation but did not have kinship.
Case 3 has an unreported missense variant c.3115C>G (p.Leu1039Val) on the NBEAL2 gene. This patient was a 34-year-old female who had severe bleeding symptoms but a mild reduction in the PLT count > 50 × 109/L. During the gene detection procedure, the platelet morphology was also examined. Degranulated platelets reminiscent of “vacuated platelets” were present on the blood smear. These pathological features are consistent with the characteristics of Gray platelet syndrome. 31,56
Cases 9 and 10 were diagnosed as BSS caused by gene variants in GPIb and GPIX, respectively. The compound heterozygous missense variants c.2T>C (p.Met1Thr) and c.901G>A (p.Glu301Lys) in case 9 were reported for the first time. The patient was a 2-year-old boy with an appearance of giant platelets on his blood smear and concomitant bleeding (mucocutaneous purpura) since early life. This patient had a significant reduction in ristocetin-induced platelet aggregation. Abnormal expression of the GPIb/IX complex was found by flow cytometry. Several analysis software programs predicted that both variants affect the primary structure of proteins. The clinical features of this patient strongly suggest the diagnosis of BSS. 57,58
Case 15 was a rare case of a variant-type GT in which the pathogenic variants were c.1022C>T (p.Ala341Val) on ITGA2B and c.1646G>C (p.Cys549Ser) on ITGB3. These 2 variants were both newly discovered and were derived from the patient’s father and mother, respectively. According to Nurden’s report, 59 the primary feature of variant GT is an unobvious reduction of the number of GPIIB/IIIa receptor molecules on the surface of platelets; however, the platelet aggregation function is defective, and some patients have reductions in their platelet counts. This patient was a 29-year-old male with long-term bleeding symptoms and a mildly low platelet count. He exhibited a near normal amount of αIIbβ3 complex, which corresponded to approximately 55% to 70% of the control levels through 3 tests. Adenosine diphosphate-induced platelet aggregation in this patient was approximately 40% lower than that in the healthy control. The 2 variants detected were located near the binding domain between GPIIB and IIIa 36,37 and might cause spatial structure changes of the GPIIB/IIIa complex, which might increase platelet clearance in the spleen and thus contribute to the reduction of the platelet count.
The other newly discovered pathogenic gene variant was identified in case 7, which was a heterozygous missense c.97T>C (p.Trp33Phe) on the MYH9 gene. MYH9-RD is a type of inherited thrombocytopenia disease caused by MYH9 gene mutations. It is also a type of macrothrombocytopenia with a higher incidence than other types. 60 MYH9-RD variants were detected in 3 of the 43 cases in our study. The patient with variant c.97T>C (p.Trp33Phe) in the MYH9 gene was a 3-year-old boy with mild bleeding symptoms since early life. Before gene detection was performed, this patient had been previously misdiagnosed with ITP and received inappropriate treatment. The patient with the variant c.2105G>A (p.Arg702His; case 5) was a 48-year-old woman with repeated petechiae and ecchymosis. She was also misdiagnosed with ITP for an extended period and presented proteins and occult blood in urine for the past 5 months. The characteristics of kidney damage initially caused the clinician to suspect that the patient had a kidney autoimmune disease. However, the related immunological tests did not support this hypothesis. Finally, the NGS results explained the cause of thrombocytopenia and kidney damage in this patient. The other MYH9-RD gene variant (case 6) was c.4269G>A (p.Asp1424Asn), which is a hotspot variant of MYH9-RD. 61 The patient was a 7-year-old boy whose major bleeding symptom was epistaxis. Before his variant in the MYH9 gene was detected by NGS, a blood smear examination was also performed, which showed inclusion bodies in some neutrophils. This cellular feature suggests to clinicians the possibility of MYH9-RD. The results of the gene sequencing provided molecular evidence for the final diagnosis.
Among the 15 cases of pathogenic variants, 8 were autosomal recessive inheritance, including 4 homozygous and 4 compound heterozygous variants. The percentage of compound heterozygous cases was higher than that reported in the literature. 62,63 This phenomenon indicated that autosomal recessive inheritance caused by homozygous variations has become rare, as consanguineous marriage in China has been greatly reduced; therefore, the majority of recessive hereditary diseases are from compound heterozygous gene variations caused by random marriage within a large population. The findings also suggested that because hereditary diseases caused by the inheritance of compound heterozygous mutations lack a familial history of hereditary diseases (the same symptom usually is not present in the family), these diseases are more difficult to identify using conventional diagnostic procedures.
Overall, among the 15 cases of diagnosed patients with inherited thrombocytopenia, 9 had been misdiagnosed with ITP, and 7 patients had been given ineffective immunosuppressive therapy. The above phenomena indicated the lack of effective preliminary screening methods for inherited thrombocytopenia. As treatments for inherited thrombocytopenia and ITP are greatly different, the therapy strategies could be changed after confirmation of the diagnosis. 3,64 Seven patients diagnosed in this study stopped their immunosuppressive therapy, and 3 were suggested to be under observation only without receiving any treatment because their reductions in the PLT counts > 50 × 109/L were mild. In addition, any drug that might influence platelet function should be avoided. Another 3 patients who had moderately low PLT counts: 20 × 109/L-50 × 109/L) were administered oral thrombopoietin therapy.
Conclusion
Because of the wide use of NGS due to its advantages of high throughput and high efficiency, more inherited thrombocytopenia-associated gene variants have been discovered. It will become an important complement for first-line diagnosis methods such as morphology and flow cytometry assessments. Furthermore, implementing NGS in routine tests will elicit a more accurate diagnosis in patients with suspected inherited thrombocytopenia.
Footnotes
Authors’ Note
Qi Wang and Lijuan Cao contributed equally to the study. Qi Wang and Lijuan Cao contributed to study design, data acquisition, and manuscript preparation; Guangying Sheng contributed to data acquisition and interpretation. Hongjie Shen, Jundan Xie, Zhenni Ma, Hong Yao, and Ma Liang contributed to gene sequencing; Lijuan Cao, Jing Lin, Ziqiang Yu, and Zhaoyue Wang contributed to collecting patients’ clinical data; Yiming Zhao, Suning Chen, Changgeng Ruan, and Lijun Xia contributed to study design; Miao Jiang contributed to study design, data analysis and interpretation, and manuscript preparation.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.