
Abstract
Coagulation disorders can be classified into 2 types, namely, type I and type II. In the former, there is a concomitant decrease in factor activity and antigen (activity–antigen ratio is 1), whereas in the latter, there is a discrepancy between factor activity which is always low and antigen which is normal or near normal (activity–antigen ratio is <1, eg, 0.5). Recently, several gain-of-function disorders have been described. These are characterized by an increased activity with respect to the antigen level. The condition involves polymorphisms of factor V and factor II, factor IX, von Willebrand disease, thrombomodulin, tissue factor pathway inhibitor, and thrombin activatable fibrinolysis inhibitor. The conditions could be subdivided into prothrombotic and prohemorrhagic. They should also be distinguished as cases of true gain of function (intrinsic increase activity without concomitant increase in protein level) and of “pseudo” gain of function (increase in both activity and protein level). This is a new concept of coagulation defects that has considerably enhanced our knowledge of blood coagulation and that should be familiar to all those interested in the mechanism of blood clotting and its disorders.
Introduction
By theoretical definition, a mutation that confers a new or an enhanced activity to a protein is maintained to be a gain-of-function mutation. However, in clinical practice, a gain of function of a clotting protein could be observed in 2 conditions, namely, (1) an increased activity due to an intrinsic structural change in the clotting protein without a similar increase in the level of the protein and (2) an increase in the level of the protein with consequent increase in its activity.
In order to respect Semantics, the former should be called “true” cases of gain-of-function disorders, whereas the second group could be termed “pseudo” gain-of-function conditions.
In practical terms, no distinctions are made, and such conditions are lumped together as “gain-of-functions disorders,” regardless of the presence of a discrepancy in the activity versus protein ratio. This is a new chapter on coagulation disorders which so far has never been dealt with in a systemic pattern, despite the fact that the first observations go back to at least 2 decades.
This is surprising, since the occurrence in humans of clotting proteins which show an increased activity disproportionate or proportionate to their size or volume is a very important finding. The purpose of the present review is to report, classify, and discuss the proven cases of clotting conditions characterized by a gain-of-function activity.
Classification
This gain of function may be manifested by an increased activity of a clotting protein or by a resistance of the mutated clotting protein (eg, abnormal prothrombins or factor V [FV] Leiden) against the inhibitors that are commonly deputed to neutralize them, namely, in this case, antithrombin and protein C. Gain-of-function disorders have been demonstrated for 2 von Willebrand (vW) disease subtypes (vWD 2B) and platelet vWD, 2 polymorphisms (FV Leiden) and prothrombin polymorphism G to A 20210, a few prothrombin abnormalities, factor IX Padua, East Texas clotting Defect (short FV disorder), and thrombomodulin (TM) abnormalities. They could be divided into prothrombotic and prohemorrhagic conditions. These conditions should be dealt with separately.
Methods and Classification
Methodology was based on a time unlimited 2 PubMed searches that were carried out on February 2016 and February 2017. Several key words including the medical subject headings (MeSH) supplied by PubMed were used. Furthermore, side tables were also examined. Original papers were obtained through the courtesy and collaboration of the Pinali Medical Library of our University. Finally, cross checking of the references listed at the end of each article was carried out, independently, by 2 of us. Gain-of-function conditions could be divided into prothrombotic and prohemorrhagic, and therefore, for the sake of clarity, they will be dealt with separately.
Prothrombotic Conditions
Factor V Leiden
The first example of gain-of-function defect is the one that causes activated protein C resistance. The FV mutation responsible for the abnormality was found in 1994 (FV Leiden), 1 but the concept of protein C resistance had been suggested 1 year before by Dahlback et al. 2
It was subsequently demonstrated that the polymorphism was diffuse all over the world with different levels of prevalence among different geographical areas (rare in Asia and frequent in Europe). This is a case of true gain of function, since the mutation in FV confers an increased activity, namely, its resistance to the inactivation by activated protein C without a concomitant increase in the protein level. The condition is associated with an increased risk of venous thrombosis, whereas its effect on arterial thrombosis is doubtful, if any.
Prothrombin polymorphism
Prothrombin polymorphism G20210A is commonly cited as an example of a gain-of-function defect. This is not completely correct since such polymorphism is associated with an increase in prothrombin antigen level. 3,4 Therefore, the ratio of activity versus antigen is always around 1, and this indicates that the prothrombin is not intrinsically hyperactive, but it is hyperactive because the protein is also increased.
This condition, contrary to what occurs with FV Leiden, could be termed as secondary gain of function to distinguish it from primary gain of function typical of the conditions where the clotting protein is not increased in quantity but is intrinsically hyperactive. A suitable term could be “pseudo gain of function.”
Since no such distinction is made in the literature, the prothrombin G20210A polymorphism is commonly quoted as a case of gain of function. The condition is known to be associated with an increased incidence of venous thrombosis, although the defect seems less severe as compared with the other known thrombophilic conditions. However, due to its diffusion (2%-3% of the European population), it plays an important role. The condition, as the FV Leiden, is rare in the Asiatic populations.
Thrombophilic abnormalities of prothrombin (antithrombin resistance)
Congenital prothrombin deficiency is one of the rarest coagulation disorders. 5,6 Homozygotes or compound heterozygotes with factor II (FII) levels of <10% of normal present a severe bleeding tendency. 5 Complete absence of prothrombin seems incompatible with life. Heterozygotes with FII levels around 40% to 60% of normal may present occasional bleeding during surgery or tooth extractions. 5 No thrombotic event has ever been reported in prothrombin “true” deficiency. 7 Recently, a few cases of prothrombin abnormalities have been associated with a thrombotic tendency. 8 –12 These abnormal prothrombins show a gain of function toward antithrombin (AT). Antithrombin is a small glycoprotein with a molecular weight of 58 000 dalton produced by the liver and circulating at a concentration of about 0.12 mg/mL. When coupled with heparin, it exerts mainly an anti-FII and anti-FX activity. Without heparin, the activity of antithrombin is markedly reduced. The increased resistance of these abnormal prothrombins (thrombins) to the action of AT creates a condition of prolonged thrombin activity that may cause thrombosis. The condition has been termed “antithrombin resistance.” 8
This is a new clinical entity characterized by a relative decrease in antithrombin activity due to the presence of abnormally resistant prothrombins (thrombins) which show a gain of function and have mutations in a special region of the molecule, encoded by exon 12, that is supposed to interact with AT. Due to these mutations, the generation of the complex Thrombin–AT (T-AT) is defective whereby antithrombin activity is decreased, thrombin persists in the circulation, and a thrombophilic state ensues. 8 –12
The first prothrombin abnormality responsible for this effect was reported in 2012 (Prothrombin Yukuhashi). 8 Subsequently, other similar cases were published in Serbia, India, and Italy. 9 –12 The 4 families involve 3 different mutations on the same amino acid, an Arginine: Prothrombin Yukuhashi Arg596Leu, 8 Prothrombin Belgrade Arg596Gln, 9 and Prothrombin Padua 2, Arg596Trp. 12 Another prothrombin abnormality seen in Japan is identical to Prothrombin Belgrade, namely, Arg596Gln. 10 All these patients showed deep vein thrombosis in young age. Prothrombin Amrita (India) is similar to Prothrombin Belgrade, but no family study is reported and, furthermore, venous thrombosis occurred at the age of 60. 11 Prothrombin Greenville shows an Arg517Gln mutation and a mild bleeding tendency but no thrombosis. 13 It is still unknown at what level of the codon sequence, a mutation can cause the shifting of a bleeding condition into a thrombophilic one. It is interesting to note that all these patients are heterozygotes for the mutation. This indirectly confirms the fact that homozygosis for prothrombin defects are very severe and sometimes incompatible with life. 5,6
The main laboratory and clinical features of these patients are gathered in Table 1. All patients had venous thrombosis; there is no information about arterial thrombosis. It is likely that other cases will be discovered. A clinical suspicion should arise from the following observations: (1) venous thrombosis in a young patient known to have no other prothrombotic defect (AT and protein C, deficiencies); (2) slightly decreased or borderline low prothrombin activity level; (3) prothrombin antigen level higher than the activity counterpart; and (4) positive family history for venous thrombosis. Needless to say that, genetic analysis is needed to confirm the suspicion.
Cases of Prothrombin Abnormalities Associated With a Gain of Function Toward Antithrombin and Therefore Cause a Thrombophilic State.a
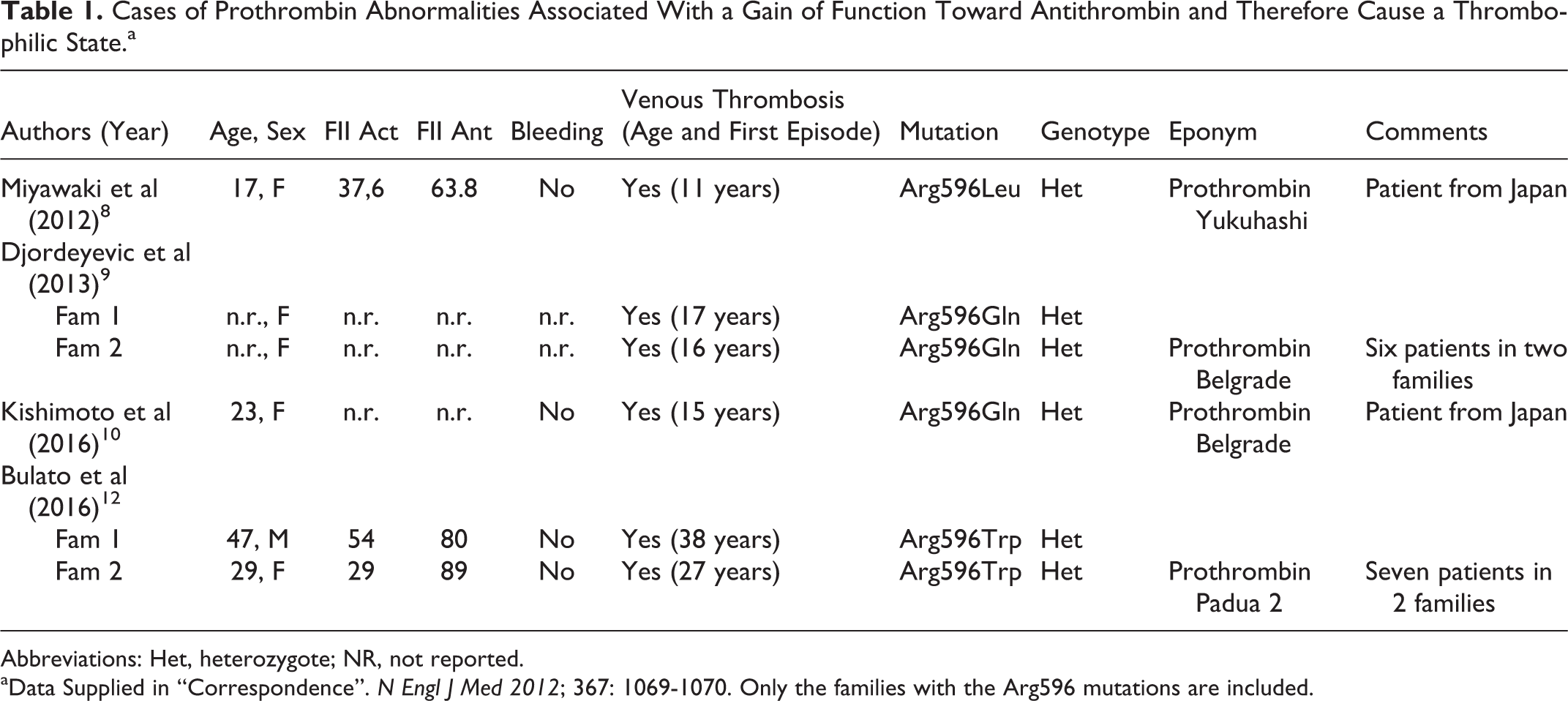
Abbreviations: Het, heterozygote; NR, not reported.
aData Supplied in “Correspondence”. N Engl J Med 2012; 367: 1069-1070. Only the families with the Arg596 mutations are included.
The occurrence of thrombosis at a young age is of paramount importance. The mean age, excluding the case from India, is 20.1 (range 11-38). The patient from India had a venous thrombosis at the age of 60. 11
It is worth noting that so far no abnormality of FX has been described which could generate an impairment in the activity of AT with a consequent AT resistance. On theoretical grounds, the possibility that such mutation in the FX gene exists is fully plausible and even likely.
Abnormal FIX (factor IX Padua)
An abnormal FIX characterized by an elevated FIX clotting activity in the presence of normal or only borderline increased antigen was described in a patient with venous thrombosis. The defect is X-linked and has been named FIX Padua. 14 The hemizygous mutation found in this patient is Arg338Lys in exon 8; a brother is similarly affected but is asymptomatic, whereas the mother is a carrier and also asymptomatic. 14
The main feature of the condition is a 10-fold increase in FIX clotting activity. This discovery prompted researchers to try and produce chimeric FIX Padua concentrates for potential use in the treatment of patients with hemophilia B. 15 –17 Several studies are in process.
Epidemiological studies in groups of patients with venous thrombosis failed to discover other cases with this FIX abnormality, indicating that the defect is rare 18,19 and probably contributes only a little to the understanding of the defect underlying the group of still unexplained familiar venous thromboses.
Prohemorrhagic Conditions
von Willebrand disease type 2B
von Willebrand disease is the most frequent bleeding disorder encountered in clinical practice. It is a complex, usually autosomal dominant, disorder that has undergone several classifications as research progressed. The vWD 2B is a rare subtype that is characterized by mutations in the von Willebrand Factor (vWF)-A1 domain. Such mutations cause a gain of function with regard to their binding to the platelet glycoprotein GPIba. As a consequence of the excessive binding, there is a disappearance or a sharp reduction in the large vWF multimers circulating in the plasma. Platelet number may also be slightly decreased. The bleeding manifestations are variable but usually severe. 20,21 From a laboratory point of view, there is an increased responsiveness or sensitivity to Ristocitin.
Platelet type VWD
This is also a rare variant of vWD. It is due to mutations in the GPIba that have been associated with an increased affinity (gain of function) for vWF. 22 The clinical picture is similar to that of vWD 2B. In other words, the condition is the specular image of the vWD type 2B. Regardless of the site of the mutation, vWF itself or its platelet receptor GPIba, the end result is similar. Platelets in this form, contrary to what occurs in vWD Type 2B, show an increased sensitivity to bovine plasma or bovine vWF.
East Texas bleeding disorder (short FV abnormality)
In 2001, a mild autosomal dominant bleeding condition was reported in a large kindred from East Texas. The patients had a mild bleeding tendency, but all clotting factors were normal, and no diagnosis was reached. 23,24 The nature of the defect was clarified several years later. 25,26 It was due to the presence of a short FV that lacked amino acids (aa) 756 to 14580 of the B domain. This short FV has a great avidity for tissue factor pathway inhibitor (TFPI). As a consequence, levels of TFPI in these patients are about 10 times greater than in normal patients. The high level of TFPI retards or inhibits the function of the tissue factor–FVII complex and, indirectly, of FX activation, and this explains the mild bleeding tendency. The condition may be envisaged as a combined FVII+FX defect due to a multifunction of a FV molecular defect. The condition is also known as “short FV defect.” 25,26
Since the B domain is not required for FV activity, this explains the normal levels of FV present in these patients. 25,26
However, the B domain is essential in maintaining FV in an inactivated state. Once it is removed or reduced, the resulting FV shows a great avidity for TFPI. 25,26 The gain of function of this abnormal FV refers to its binding to TFPI. This leads to the prolongation of TFPI life and to its increased plasma levels with consequent bleeding tendency. In other words, the high TFPI levels interfere with the aFVII-tissue factor formation and activity with a consequent appearance of a hypocoagulable state.
Recently, another FV defect was reported (FV Amsterdam). 27 This is also a “short FV,” but it seems different from East Texas FV in that only 63 aa. of the B domain are missing. The number of missing aa. in FV East Texas is 702. In this case, the deletion of most of the B domain results in a greatly increased binding of TFPI. In the case of FV Amsterdam, the increase in TFPI is less pronounced, 25,26 but the clinical and laboratory picture is similar to that of East Texas FV.
It is conceivable that new cases will be discovered. From a diagnostic point of view, the most important suspicion should arise when a mild bleeding tendency is not explained by any known defect, even if a rare one as antiplasmin deficiency. Other important clues are the positive family history for a mild bleeding tendency. The suspicion has to be confirmed by the finding of high levels of TFPI and the correct diagnosis clinched by genetic studies. The significance of an elevated level of TFPI had never been suspected to play a role in the diagnosis of a bleeding tendency in humans. 28 TFPI is a polypeptide that has 2 isoforms TFPIα and TFPIβ which show different inhibitory activities. TFPI congenital deficiency has never been described in humans, but the inhibitor is at present time the object of great scientific interest. 28
Thrombomodulin abnormality–associated gain of function
Thrombomodulin is a transmembrane glycoprotein composed of 557 aa. It has a great affinity for binding to thrombin and to transform it from a coagulant to an anticoagulant compound. 29 The complex thrombin–thrombomodulin (T-TM) has 2 main actions: (1) It activates protein C which by its turn, once activated, downregulates aFV and aFVIII and (2) it also activates the thrombin activatable fibrinolysis inhibitor (TAFI). 29 Activated TAFI inhibits the adherence of plasminogen to the surface of the fibrin clot, thereby decreasing the effect of plasmin-induced fibrinolysis. As a consequence, TM plays an important role in regulating the structure and resistance of the fibrin clot. Congenital TM deficiency has never been described. In 2014 and 2015, two families with a mild bleeding tendency but normal coagulation tests were reported. 30,31 Extensive and elegant investigations demonstrated that the defect was due to greatly elevated TM levels. Genetic analysis of the TM gene showed the presence of the same mutation, namely, Cys537Stop in both families. 30,31 Such mutation is responsible for the gain-of-function effect of TM.
The greatly increased levels of TM were maintained to be responsible for the bleeding tendency. Bleeding is usually mild, but it may become severe after surgical procedures (generation of tissue factor). 31
The exact mechanism of bleeding is still not fully explained, but it seems that in these patients, the effect of the T-TM complex on FV and FVIII (potentially hemorrhagic action) prevails on the effect on TAFI (potentially procoagulant action). 30,31 These patients also show a decreased prothrombin consumption which has not been explained. 30,31
The description of these 2 families with the same mutation has considerably spurred the interest in the T-TM complex activities. 32,33 Further studies are needed to clarify the exact features of the defect.
Hemophilia BM
Since by definition a gain of function of a coagulation protein is characterized either by an increased activity or by the appearance of a new activity, hemophilia BM should also be included in this review. Hemophilia BM was first described in 1967. 34 The defect is a Hemophilia B+ or a cross-reactive material positive form. The peculiarity resides in the fact that the FIX antigen is not inert but has acquired an anticoagulant or inhibitor activity toward tissue thromboplastins (TT). Such new inhibitory activity becomes particularly evident when slow TT like those obtained from bovine brain 35,36 are used. This is a new function, and therefore the condition should be included. The main diagnostic feature is a sharp prolongation of PT when a bovine brain thromboplastin is used together with a prolonged aPTT. The latter is corrected by a mixture time with normal plasma, whereas the former is not. Several mutations have been detected in the activation site or in the catalytic region of FIX. 36,37 Surprisingly, the condition is often ignored. For example, it is not listed in the index of Colman’s Textbook of Thrombosis and Hemostasis (Fifth edition).
Discussion and Conclusions
We have been accustomed to the study of mainly 2 types of clotting factor deficiencies, namely, type 1 and type 2.
The former defects are characterized by an equivalent decrease in activity and antigen of a given clotting protein and are defined as cases of “true” deficiency. In the latter, there is a discrepancy between activity that is always low and antigen that is normal or near normal. On theoretical grounds, the gain-of-function defects could be defined as type III, since the reverse of what is seen in type II usually occurs, namely, a highly increased activity in the presence of normal or variably increased antigen.
In other words, the ratio of activity versus antigen, in a theoretical example, results to be 1, 0.5, or 1.5 for the type I, type II, and type III defects, respectively. These would be the cases of “true” gain of function. However, the concept has been extended even to cases in which there is an increase in function due to an increase in antigen or protein. The latter form could be defined, more properly, as “pseudo” gain of function. There is an increased activity or function because there is also an increase in the antigen or protein level. The activity–antigen ratio always remains 1. Matters may be complicated by the fact that activity or antigen assay methods are not yet always satisfactory for some clotting proteins (eg, TM).
The number of type III or gain-of-function defects is still limited, but it is likely that further studies will discover other cases.
It is likely, for example, that, due to similarities with prothrombin, an abnormal FX could be discovered with a mutation in the area which interferes and is supposed to be neutralized by AT will be reported. The resulting effect will be a gain of function of the abnormal FX with consequent AT resistance similar to what is seen for the abnormal prothrombins.
The same is true for FVIII since it is known that high FVIII levels have been reported to be associated with thrombosis. 38 Again, it is likely that a mutation in the large molecule of FVIII could be responsible for a gain of function and the thrombotic tendency. FVIII, together with FV, is neutralized by activated protein C. Families with elevated levels of FVIII activity and thrombosis have been described, but, so far, no explanation has ever been obtained. 39
Since it has been demonstrated that a mutation in FV (FV Leiden) can cause activated PC resistance with the consequent appearance of a thrombophilic state, one could also suspect the existence of an FVIII-derived activated protein C resistance.
Furthermore, could a mutation in protein C cause a defective inactivation of FVIII and/or FV with their longer persistence in the circulation?
The above-mentioned examples refer all to thrombophilic conditions. The “short FV” abnormality or East Texas bleeding disorder refers instead to hemorrhagic conditions. This indicates that gain-of-function defects may be involved in both prothrombotic and prohemorrhagic conditions. The avidity of these “short FVs” for the TFPI has demonstrated the complexity of the clotting system. Nobody could have ever thought that TFPI could be involved in a bleeding tendency.
Since TFPI is a protein and is therefore dependent on a gene function, it is likely that its congenital deficiency will be demonstrated in the future. The recent description in 2 patients, that a mutation in the TM molecule is associated with a mild bleeding tendency, is in agreement with these considerations. Further studies will certainly clarify all these possibilities and contribute to the explanation of an ever-enlarging number of the so called idiopathic thromboses and clarify peculiar bleeding defects. These studies once again demonstrate the importance of patients in the understanding of the clotting system. 40 –42
They also indicate that blood coagulation has become such a vast field of investigation and has become so overspecialized that it is rare that one single author could grasp the connections, among the different phases, or aspects of it. The reviewers usually deal with one subject, namely, vWD, fibrinolysis, FVIII, fibrinogen, and so on, never or seldom tackle general principles and mechanisms involving several coagulation proteins. In the past, when blood coagulation was thought to be relatively simple, coagulation experts were common. Today, they have become a rare species. This may explain, at least in part, the fact that the gain-of-function conditions have never been, so far, the object of an overall review.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.