
Abstract
Factor VII (FVII) deficiency is a rare inheritable bleeding disorder affecting 1/500 000 individuals. Clinical manifestations are heterogeneous, from asymptomatic to severe and potentially fatal bleeding. These clinical manifestations do not correlate well with FVII plasma levels. For this reason, FVII-deficient patient management during surgery or for long-term prophylaxis remains challenging. Laboratory testing for FVII activity is, however, the first-line method for FVII deficiency diagnosis and is helpful for managing patients in combination with clinical history. Additional testing consists of FVII immunoassay and genetic testing. Genetic abnormalities on the FVII gene are heterogeneous and can translate into quantitative or qualitative defects. Some of the latter can react differently with different thromboplastins; this can be misleading for the laboratory as no consensus exists at present on an FVII deficiency diagnosis methodology. Indeed, no single test is able to predict accurately the bleeding risk. This review provides a broad picture of inherited and acquired FVII deficiency with a particular focus on laboratory diagnosis.
Keywords
Introduction
Factor VII (FVII) also known as proconvertin is a clotting factor of the human coagulation system. This free circulating glycoprotein belongs to the serine protease family. It is a vitamin K-dependent protein that is synthetized exclusively by the liver. Plasma levels range around 0.35 to 0.60 mg/L (for a normal coagulant activity comprised between 70% and 140%), which is 10 times less than other vitamin K-dependent factors. Its half-life is extremely short (4-6 hours). Factor VII is the only clotting factor that has a small proportion (1%-3%) of free circulating activated form (FVIIa) in the absence of coagulation activation. 1,2
Factor VIIa by itself is not sufficient to initiate coagulation; it requires the presence of tissue factor (TF). Following injury, the integral membrane protein TF is exposed into the vascular lumen and can bind the free circulating FVIIa in order to trigger coagulation. The action of the FVIIa–TF complex generates a burst of activated factors IX (FIXa) and X (FXa), which results in the formation of stable fibrin clot. 2
Tissue factor and FVIIa interaction involves all domains of FVIIa, that is, the catalytic domain, the Gla (gamma-carboxyglutamic acid) domain, and the 2 epidermal growth factor (EGF)-like domains. The close bond of TF to FVIIa induces allosteric activation of FVIIa by enhancement of the enzymatic activity, thus initiating blood coagulation by the activation of factor X and factor IX. 3 Only traces of FVIIa bound to TF are required to trigger the whole mechanism of coagulation.
Cross-species compatibility of FVIIa with TF from animal origins has been widely investigated in an attempt to propose surrogate reagents to human tissue thromboplastin that was used in the very first time of coagulation assays. 4 At present, human recombinant thromboplastin is available, but thromboplastin from rabbit brain (and from an ox brain in a lesser extent) is still broadly used. The various sources of TF found in the available thromboplastins may impact the sensitivity of reagent to coagulation defects including FVII polymorphisms.
Functional testing is a key to the routine diagnosis of FVII deficiency. It is based on the results of routine assays (prothrombin time [PT] and activated partial thromboplastin time [aPTT]) and the specific measurement of FVII coagulant activity (FVII:C). In this review, we present the different aspects of FVII deficiency: what is it, how to diagnose it, what the clinical manifestations are, and how to manage patients.
Factor VII Deficiency
Seventy percent of FVII activity is usually considered as the lower limit of the reference range. This lower limit value has been based on a statistical approach of results obtained of a reference sample group reflecting the normal population. However, clinical manifestations rarely appear when FVII:C is above 30%. 5 This value can therefore be considered as the cutoff for clinical manifestations. Caution should be exercised regarding the reagents used for PT and FVII activity measurements as FVII deficiency reagent sensitivity varies across reagents (see Laboratory Diagnosis of FVII deficiency section). Factor VII deficiency can be caused by a genetic impairment or can be acquired, secondarily to a pathological situation.
Acquired FVII Deficiency
Acquired FVII deficiency can be isolated or combined with other decreased coagulation factor levels. Only a few cases of isolated acquired FVII deficiency are reported in the literature, even if some authors suggest that this kind of defect is underestimated. 6,7
However, decreased FVII activity is a nonrare situation usually encountered in the course of the exploration of a prolonged PT. Several pathological settings associated with coagulation disorders can lead to low coagulation factor levels. Because the half-life of FVII is the shortest, a pseudo-isolated FVII defect can be seen before other factor levels decrease. The pathological mechanisms leading to a combined decline of coagulation factors are: A decrease in the synthesis, for example, associated with liver failure. All the coagulation factor levels and the one of their inhibitors are decreased. A defective synthesis, for example, hypovitaminosis K syndrome caused by insufficient intake, malabsorption, or antivitamin K medications. Only vitamin-K dependent factors (II, VII, IX, and X) and protein C and protein S levels are decreased. Consumption syndrome, for example, hyperfibrinolysis or disseminated intravascular coagulation.
Isolated acquired FVII deficiency is extremely rare. When identified, they are mostly related to malignancy and most of the time linked to the presence of an inhibiting antibody. Few cases have been described in major trauma, postsurgery, sepsis, aplastic anemia, or bone marrow transplantation. 7,8 Clinical outcomes are variable and could be potentially severe. 9 Mechanisms involved have not been fully explained. On theoretical grounds, acquired FVII deficiency could be due to an abnormal or decreased synthesis, an accelerated consumption or catabolism, a neutralization by an antibody, or an abnormal absorption by tumor mass. The release of a massive bulk of TF in the plasma during a severe trauma or chemotherapy can be associated with an accelerated consumption of circulating FVII.
Inherited FVII Deficiency
Factor VII gene, located on chromosome 13q34, spans about 12 800 bases and comprises 9 exons preceded by a promoter region. 10 The transcripted protein circulating in the plasma is a 406-amino acid single chain of 50 kDa. When activated, FVII protein is cleaved at the Arg212-Ile213 liaison into 2 chains: the heavy chain, which contains the catalytic domain, and the light chain, which contains the γ-carboxyglutamic acid (Gla) and 2 EGF-like domains. 11 Gene and proteins are structurally homologous to other coagulation serine proteases, particularly factor IX, factor X, and protein C.
Congenital FVII deficiency (OMIM 227500) is the most common among the rare congenital bleeding disorders and represent one-third of them. The pattern of inheritance is autosomal recessive. Global prevalence is 1/500 000, 12,13 but it can be more frequent in parts of the world where consanguinity is more frequent. The prevalence of FVII deficiency may be underestimated because of the possibility of asymptomatic patients even if their FVII:C is decreased.
The spectrum of mutations is wide. To date, more than 250 different mutations scattered along the FVII gene have been identified. 14 An FVII mutation database is available at http://www.hgmd.cf.ac.uk/ac/gene.php?gene=F7. 15 Mutation types are heterogeneous: missenses for 70% to 80% of cases, frequent splicing site changes, rare nonsense, and small deletions. 12,16 Association of 2 different mutations is relatively frequent. Homozygous or compound heterozygous mutations can translate into the same clinical phenotypes associated with moderate to severe bleeding. However, patients with homozygous or compound heterozygous mutations are indistinguishable unless genomic assays are performed. Furthermore, diversity in the clinical phenotypes has been observed across patients with identical genotypes. 14
Among the diversity of genetic impairments of FVII-deficient patients, some mutations have been identified to be associated with different sensitivities of FVII:C toward thromboplastins from different origins. These special variants, named FVII Padua, FVII Nagoya, and FVII Tondabayashi, will be addressed further in this review.
Laboratory Diagnosis of FVII Deficiency
Three types of circumstances can lead to the diagnosis of FVII deficiency: laboratory workup performed in the case of bleeding events, screening in the context of family history of FVII deficiency (when at least 1 carrier of the mutation is known in the family), or a serendipitous finding. The median age for the discovery of an inherited defect is 8 years. However, when the defect is severe, bleeding manifestations occur during the first month of life.
Factor VII deficiency can be easily suspected when a hemostasis workup reveals an isolated prolongation of PT with a normal aPTT (Table 1). The prolonged PT is corrected by the addition of normal pool plasma (mixing study: 1 volume of test plasma + 1 volume of normal pool plasma). Factor VII deficiency is usually characterized by an FVII:C below 70% (0.7 IU/mL), even if clinically relevant manifestations mainly appear when FVII:C is <30% (clinical manifestation threshold).
Expected Results of Global and Specialized Laboratory Assays for the Diagnosis of FVII Deficiency.
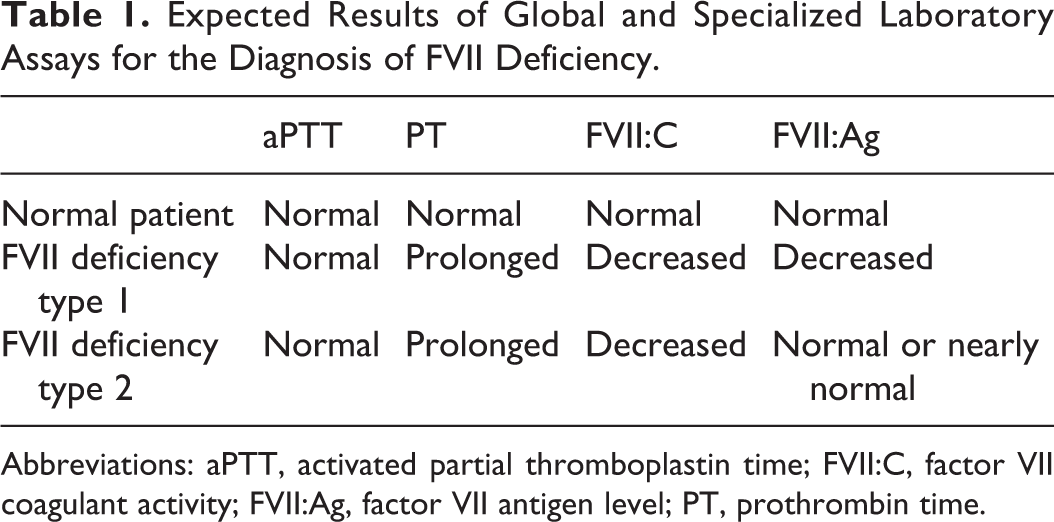
Abbreviations: aPTT, activated partial thromboplastin time; FVII:C, factor VII coagulant activity; FVII:Ag, factor VII antigen level; PT, prothrombin time.
In order to rule out a transient or a combined defect, a sample should be drawn a couple of weeks after the first determination, and testing should be repeated to confirm the congenital deficiency.
Factor VII congenital deficiency is commonly divided into 2 groups, type I, which are quantitative defects, and type II, which are qualitative defects. The former are characterized by a concomitant decrease in FVII activity and antigen levels. In the latter, there is a clear discrepancy between FVII activity, which is always low, and antigen, which is normal or nearly normal (Table 1).
Preanalytical Conditions for FVII Measurement
The 1-stage assay of FVII:C is derived from the PT assay and can be affected by many preanalytical and analytical variables. Common preanalytical conditions for hemostasis tests are described by the Clinical and Laboratory Standard Institute (CLSI) H21-A5 document. These recommendations should be cautiously followed
17
: Sample collection has to be done on 0.109/0.105 M (3.2% vol/vol) trisodium citrate blood collection tubes (1 volume of citrate solution:9 volumes of whole blood). Tubes should be mixed immediately after blood collection by gentle inversions since overmixing and harsh mixing may cause hemolysis or platelet activation. Transport should be performed at room temperature and not at 4°C to 8°C as cold exposure can promote the activation of FVII, leading to substantial overestimation of the true FVII level.
18
Tubes should be centrifuged at 1500g for no less than 15 minutes. Higher speed and shorter duration such as 2000g for no less than 10 minutes may also be used.
17
Centrifugation should be performed at room temperature to avoid FVII cold activation. Neither blood nor plasma samples for determination of FVII:C should be stored at 4°C. In case of delayed testing, samples should be immediately frozen and stored at −20°C or below (for short-term storage up to 2 weeks) or −70°C or below (for long-term storage). Thawing should be performed rapidly in a water bath (37°C).
19
However, fresh plasma testing should be preferred for FVII:C instead of frozen sample testing, whenever possible, to avoid FVII activation by cold exposure.
Factor VII:C Clotting Assay
Factor VII activity measurement is accomplished by mixing citrated plasma sample, FVII-deficient reagent, and thromboplastin (start reagent containing TF, phospholipids, calcium chloride, and frequently a heparin neutralizer). Time for clot formation is measured after the addition of the start reagent, which initiates the clotting process. Although all the clotting factors except FVII are present in excess, clotting time is inversely proportional to the amount of FVII present in the patient sample.
Accuracy and precision of FVII:C assay depend on 3 factors: (1) sensitivity of thromboplastin used, (2) calibration materials, and (3) quality of the reagents used for the FVII:C assay.
20
At present, 3 types of commercially available thromboplastin exist. They differ by their origin (rabbit brain, ox brain, or human recombinant thromboplastin). This results in variability in PT sensitivity across reagents. Specifically, the responsiveness of FVII assay, as well as PT, varies depending on the thromboplastin used. This variability can be seen for normal plasma and to a larger extent in some rare types of qualitative FVII defects (FVII Padua, FVII Nagoya, and FVII Tondabayashi). This is discussed in the Thromboplastin Sensitivity Across Qualitative FVII Defect section. To date, certified reference materials are used by manufacturers to assure accuracy of manufactured calibrators and to ensure accurate factor assays for laboratories. Despite this, interlaboratory precision varies depending on the calibrator used, especially for FVII levels below 20%. It has been suggested that the calibrator used for FVII measurement could also contribute to the sensitivity discrepancies observed across thromboplastin reagents as many laboratories use the calibrator/thromboplastin combination provided by the same manufacturer.
21
The quality of the reagents may play an important role in the precision of FVII:C assay. Indeed, manufactured FVII-deficient plasma may have a residual FVII:C. This could lead to an overestimation, especially for low FVII levels. According to the 2016 CLSI document H48-Ed2, Determination of Coagulation Factor Activities Using the 1-Stage Clotting Assay,
19
the mean activity of deficient plasmas should contain a level of <1% of factor measured. Furthermore, thromboplastin optimal quality is also a prerequisite since Smith et al described that thromboplastin with trace contamination of FVIIa decreased sensitivity to plasma FVII:C by the activation of the plasma. This contamination could also increase sensitivity to plasma levels of FV, FX, and prothrombin.
22
Thromboplastin Sensitivity Across Qualitative FVII Defects
Although rare, some FVII dysfunctional variants display different FVII:C activities when measured with thromboplastin preparations derived from different species. 1,23
This is the case for FVII Padua, a special variant of FVII deficiency (Arg304Gln in exon 8), which has a very low prevalence but a worldwide distribution (Italy, Iran, United States, France, Japan, England, and Tunisia). It is characterized by different reactivity toward different tissue thromboplastins. 24 An ox brain thromboplastin yields normal values of FVII:C, whereas rabbit brain thromboplastin yields lower FVII:C levels. Human recombinant thromboplastin provides intermediate results.
There is full agreement that FVII Padua is a type II variant since antigen levels are always normal. The ratio FVII antigen (FVII:Ag) to rabbit brain, human tissue, and ox brain FVII:C activities are around 25, 4, and 1, respectively. 25 Girolami et al suggest the use of at least 2 thromboplastins from 2 different sources (rabbit brain vs ox brain) as a surrogate approach to molecular diagnosis to identify this peculiar defect. 25 From a clinical standpoint, results yielded with human recombinant thromboplastin in FVII Padua defect (around 30%) could better reflect the clinical phenotype as all the cases described in the literature have mild bleeding manifestations or are not symptomatic, whereas results obtained with rabbit brain thromboplastin (5%-10%) would suggest a more pronounced defect, and results obtained with ox brain thromboplastin (around 100%) would classify the patient as normal. The use of rabbit brain thromboplastin is, however, recommended as a first-line screening test in case of FVII defect suspicion because of its increased sensitivity. 25
FVII Nagoya (Arg304Trp) and FVII Tondabayashi or Shinjo (Arg79Gln), which are also type II variants, have been shown to exhibit a similar pattern of variable reactivity toward different tissue thromboplastins as FVII Padua. The occurrence of these defects is sporadic. 26
Results of FVII:C in patients bearing homozygous qualitative mutations impacting laboratory coagulation assays are reported in Table 2. The discovery and the study of such mutations impairing the TF–FVII liaison had a great influence on the understanding of molecular mechanism of FVII physiological pathway. The presence of these mutations localized among 2 different sites of the FVII gene indicates that at least 2 areas of FVII are involved in TF binding, namely an EGF-like domain on the light chain (Arginine-79) and the catalytic domain (Arginine-304). 27 In contrast, no case of quantitative FVII (type I) defect has ever been described as displaying variable reactivity toward different thromboplastins.
Factor VII:C Activity of Different Homozygous FVII Defects According to Girolami et al. 27
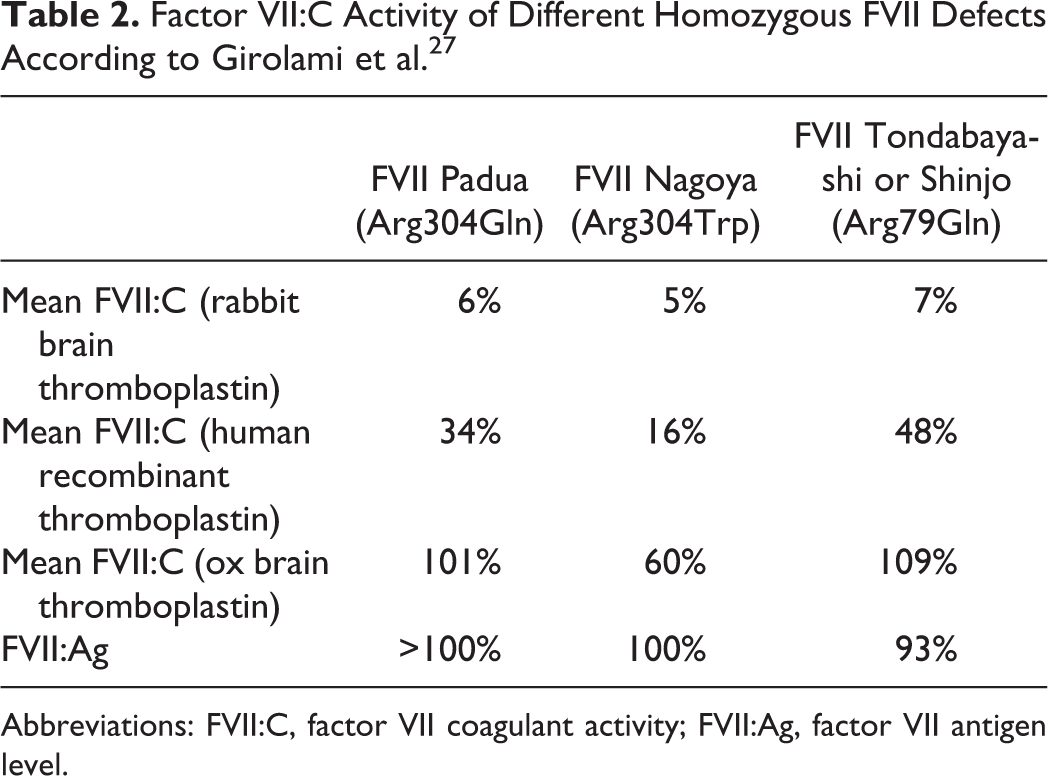
Abbreviations: FVII:C, factor VII coagulant activity; FVII:Ag, factor VII antigen level.
Factor VII Antigen Level
The determination of FVII:Ag level can be performed by enzyme-linked immunosorbent assays or immunoturbidimetric assays, using a monoclonal antibody targeting an epitope specific for the free-circulating FVII. The quantitation of the FVII antigen must not be used as a first step in the diagnosis of FVII deficiency since some qualitative defects can be missed. Moreover, the result is not predictable of a bleeding tendency. Factor VII:Ag assay is, however, essential to discriminate the quantitative defect from the dysfunctional form of FVII deficiency (type II). Finally, FVII:Ag is a necessary tool to better understand the large panel of mutational mechanisms of FVII deficiency.
Factor VIIa Activity Assay
Specific FVIIa assays have been introduced to monitor recombinant activated FVII (rFVIIa) therapy in FVII-deficient patients as well as in patients having hemophilia with inhibitors.
The method is based on the use of recombinant TF, which acts as cofactor for FVIIa but does not activate FVII. 28 The overall correlation with FVII:C is relatively good in patients receiving rFVIIa therapy. Although and because FVIIa assay has lesser variability than FVII:C, FVIIa assay may be more suitable for the monitoring of rFVIIa therapy than FVII:C. 29 However, FVIIa assay is not indicated in the diagnosis of FVII deficiency.
Genetic Testing
The FVII gene (F7) was first sequenced in 1987 and has been extensively investigated by DNA sequencing of all coding regions, exons/introns, boundaries, and the promoter regions. At present, sequencing methods enable very efficient detection of mutations in patients with FVII deficiency. Total gene sequencing is recommended as a first step for molecular diagnosis since there is no highly recurrent mutation, the length of the gene is short, and the more frequent abnormalities are single-nucleotide mutations. Direct sequencing is able to detect 90% to 92% of mutated alleles. Nonetheless, there are still 10% of FVII deficiencies in which mutations are not found. This suggests that the option of FVII deficiency caused by mutations in another gene other than FVII gene is still an open question. 30
Prenatal diagnosis of severe FVII deficiency is available but should be restricted to families who have a history of severe bleeding episodes associated with an FVII defect. Genetic analysis performed on cord blood samples is the gold standard method. In case genetic assays are not available, clotting assays on cord blood obtained by cordocentesis between the 17th and 21st weeks of gestation could be performed; however, reconfirmation is needed after birth in such case. 31,32
Acquired FVII Deficiency Diagnosis
As for congenital FVII deficiency, an isolated PT prolongation is often the first indication leading to a specific coagulation workup. Acquired combined factor deficiency is more common than acquired isolated FVII deficiency. Since FVII has the shortest half-life among the factors, its level decreases before other factor levels are affected. For that reason, the diagnosis of an acquired FVII deficiency should be considered with caution. A careful evaluation of all the clotting factors has to be performed and possibly repeated to exclude a combined acquired defect. All causes that may lead to such defects, that is, liver failure, malabsorption, or vitamin K antagonist medication should be carefully assessed. Differential diagnosis between acquired and inherited FVII defects requires a thorough family study in order to demonstrate a potential inheritable defect. Conversely, a history of a normal FVII activity assay rules out a congenital FVII deficiency in most of the cases, provided a false normal result due to the lack of reagent sensitivity has been ruled out.
The presence of an inhibitor, which has been reported in some cases of acquired FVII deficiency, can be revealed by a PT mixing study, and titration could be performed using a Bethesda-based method (serial dilutions of test plasma + normal pool plasma tested after 1-hour prolonged incubation at 37°C in a water bath).
Clinical Manifestations
Bleeding manifestations of FVII deficiency are extremely heterogeneous regarding both localization and severity. Clinical features range from asymptomatic condition to serious and potentially fatal bleeding episodes. Clinical phenotypes correlate poorly with FVII activity levels. 1,2 An FVII:C level below 2% correlates with an increased risk of severe bleeding, but it is not rare to observe the lack of spontaneous or provoked bleeding manifestations in cases of FVII:C below 1%. On the other hand, patients with FVII:C level above 5% have been reported to have a history of severe bleeding manifestations. 30 This lack of correlation between clinical and laboratory phenotypes is not clearly understood. It has been suggested that other factors than FVII such as TF, von Willebrand factor, or platelets could be involved in the expression of bleeding tendency in FVII deficiency. Unknown regulators are not either excluded. 5
The minimal level of FVII needed to avoid bleeding in different clinical situations is not known. Some authors state that an FVII:C level above 8% is associated with an extremely low risk of severe bleeding (defined as hemorrhage requiring blood transfusion or factor concentrates—including FVII concentrate infusions) during surgery. 5 Other authors report that a level of 15% to 20% of FVII:C is generally sufficient to prevent spontaneous bleeding. 12 It is mainly acknowledged that FVII plasma levels should be over 20% to control bleeding and to ensure safety during surgery. 2,33 The combined enhanced effect of TF–FVIIa complex is sufficient to trigger effective coagulation with low levels of FVII; this explains why only low FVII plasma levels are required to control bleeding during surgery.
Among the patients with congenital FVII deficiency, up to one-third of individuals are asymptomatic. In the remaining two-thirds, most of the patients experience a mild clinical picture, whereas only 10% to 15% of patients experience severe manifestations. 14,23
Clinical manifestations can be divided into nonsevere and severe bleeding categories. In the former, most frequent symptoms are epistaxis (60%), gum bleeding (34%), easy bruisability (36%), menorrhagia (69% of females), and other minor bleeding (hematomas, hematuria). These nonsevere manifestations in FVII deficiency mimic that of a platelet disorder and usually do not require any treatment. In the latter, extensive bruising and hemarthrosis are frequent (19% for both of them), gastrointestinal (GI) bleeding is relatively frequent (15%), whereas central nervous system (CNS) bleeding is less frequent (2.5%) but even more serious and life threatening (Table 3).
Clinical Manifestations of Symptomatic Patients With Factor VII Deficiency.
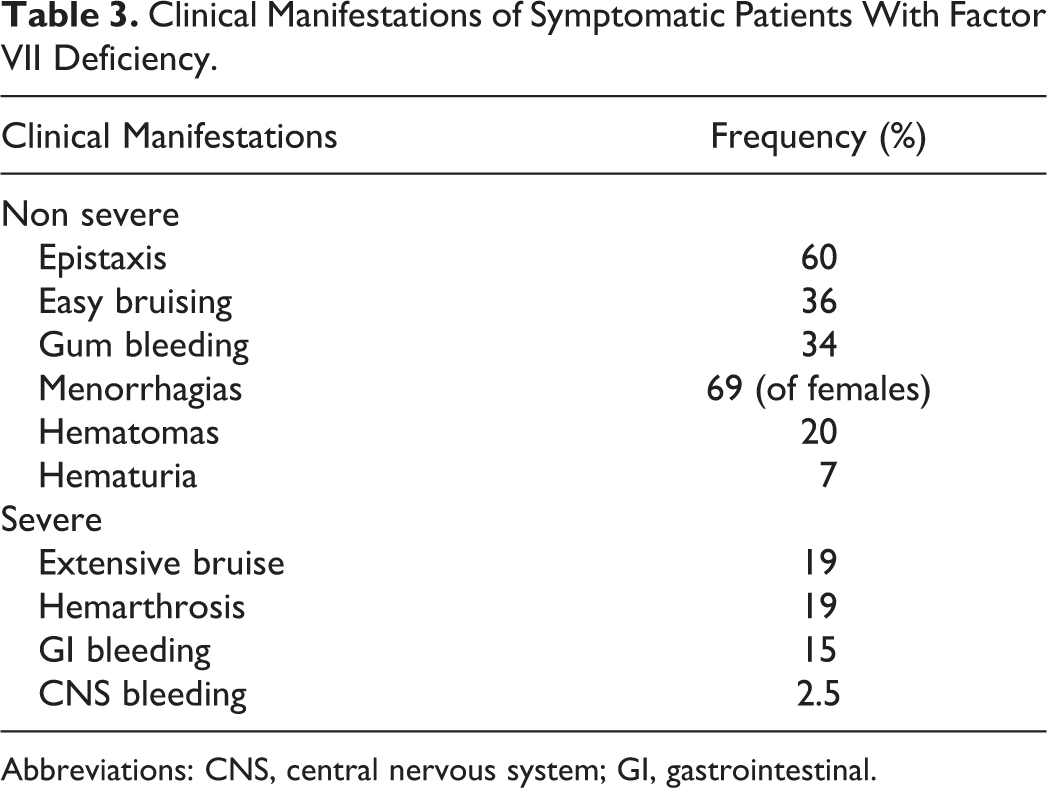
Abbreviations: CNS, central nervous system; GI, gastrointestinal.
McVey et al noticed that complete absence of FVII in plasma is usually incompatible with life, and individuals die shortly after birth due to severe hemorrhage. 30 Heterozygotes are usually characterized as clinically asymptomatic. A large study with heterozygous FVII deficiency reveals that nearly 20% of them present some minor bleedings such as epistaxis, gum bleeding, easy bruisability, or menorrhagia. No severe symptoms have ever been reported among heterozygous patients. 14
Thrombotic events (deep venous thrombosis and pulmonary embolism) have been reported in 3% to 4% of patients with FVII deficiency. 1 Venous thrombosis seems to be more frequent in FVII deficiency than arterial thrombosis. The reasons underlying these peculiar thrombotic manifestations are unknown, even though in the majority of patients associated thrombotic risk factors are present. Altogether, FVII deficiency does not ensure a sure protection from venous thromboembolisms. 34,35
The development of a FVII inhibitor in patients with FVII deficiency is a rare complication. It can be related to replacement therapy (about 1%-3% of treated patients, in most of the case in infants with a very severe phenotype) or linked to malignancy or autoimmune disease. However, no anaphylactoid complication to replacement therapy has ever been showed, even if a prophylaxis is still conducted. 36,37
Management
Acute Bleeding
Because of the wide range of clinical phenotypes and the lack of correlation between bleeding manifestations and FVII:C level nor genetic defects, bleeding risk prediction and replacement therapy strategy during surgery or acute bleeding event are real challenges for the practitioner. Moreover, unlike hemophilia A and B in which a bleeding severity classification is proposed based on the plasma level of factor VIII and factor IX, respectively, patients with FVII deficiency cannot be classified and managed only according to the level of FVII:C or any other laboratory test. This is because no conventional clotting assay has ever been proven to correctly predict the bleeding risk tendency.
At present, scientifically established procedures for perioperative replacement are lacking. As a result of the coordination of an extensive, prospective trial regarding the management of FVII deficiency, a few recommendations have been proposed by the Seven Treatment Evaluation Registry and the International Factor VII Deficiency Study Groups. 38,39 The most useful tools to evaluate the bleeding risk available up to now are the combination of the patient’s bleeding symptoms history, the surgery type (type of organs involved and type of anesthesia), and the level of FVII:C. 39
When bleeding symptoms are severe and well documented (recurrent hemarthrosis, GI, and CNS hemorrhage) or if surgery procedures carry the risk of a major or disabling bleed (eg, neurosurgical, urological, and cancer-related procedures), a decision of giving replacement therapy should be taken, unless the plasma level is above 20%.
The treatment question remains open when the surgery has a minor risk of bleeding with a positive hemorrhagic history or a major surgery with intermediate FVII:C level and a negative bleeding history. If a nonreplacement therapy decision has been made, medications must always be quickly and easily available. Caution must be exercised with children who could have a “falsely negative” hemorrhagic history because they have not yet experienced any prior bleeding challenge.
Several therapeutic options can be offered to patients regarding severe bleeding symptoms. Recombinant FVIIa is the most widely accepted replacement therapy in patients with acute bleeding symptoms. The effectiveness to control bleed is excellent, and it can be used at low doses (15-30 µg/kg of body weight) compared to the dosing of bypassing therapies in hemophilia. This can be explained by the lack of competition with endogenous FVII as it is the case in patients having hemophilia with inhibitors. Thrombosis related to rFVIIa treatment is very rare; there is no virus transmission-associated risk, and development of inhibitory antibodies against rFVIIa has been recorded rarely. The main issues with this therapy are (1) that it requires frequent bolus injections to counteract its short half-life and (2) that it is extremely expensive. 40
Monitoring of rFVIIa therapy has been proposed to be performed with PT, FVII:C, or FVIIa assays. However, it is known that FVII:C results vary greatly across laboratories (primarily due to the thromboplastin used), as do PT results, even if measured using thromboplastins of similar sensitivity. The use of FVIIa-specific assay appears to be more suitable for the monitoring of rFVIIa treatment, in patients with hemophilia as well as FVII-deficient patients. 29,41
Plasma-derived FVII concentrates may provide an alternative to rFVIIa, but experience is limited. Furthermore, availability of plasma-derived FVII concentrates is becoming uncertain in some countries.
Fresh frozen plasma (FFP) is a cheap and easily available option, but its effectiveness is limited due to the high volumes that have to be administrated. Fluid overload may be a serious complication of FFP transfusion. 42 In addition, although very unlikely, viral contamination cannot be totally excluded. Fresh frozen plasma remains an acceptable option for newborns for whom the volumes to infuse are reasonable and when it is not necessary to dispense the therapy too frequently (eg, a minimally invasive diagnostic procedure or a minor cutaneous surgery).
Prothrombin complex concentrates (PCC) and activated PCC (aPCC) could also be used but were reported to be associated with posttreatment thrombosis.
Finally, the antifibrinolytic tranexamic acid could be used in combination with replacement therapy or alone in minor bleeding symptoms such as mild menorrhagia. 23
Most of the time, acquired FVII deficiency is related to an underlying pathology, and normalization of FVII activity level is usually observed by the effective treatment of its causative pathology. Hence, the management of acquired FVII deficiency involves tackling the underlying disease. Usually, clinical manifestations are associated with mild to moderate bleeding, which may become relevant in case of surgery. Therapeutic options, in case of significant bleeding, are the same as in congenital defects (FFP, PCC, aPCC, and plasma-derived or recombinant FVIIa). 6
Prophylaxis
Long-term prophylaxis in FVII deficiency cannot be considered widely in contrast to hemophilia. This is principally due to the fact that FVII and FVIIa have very short half-lives (<3 hours). Patients eligible for long-term prophylaxis are those with a previous episode of severe bleeding such as CNS, GI bleeding, or recurrent hemarthrosis. Prophylaxis is generally initiated soon after the first severe bleeding episode (ie, majority of patients are newborns). 12 The most commonly used dose regimen yielding “excellent” outcomes is a weekly dose of 90 µg·kg−1 of rFVIIa split into 2 or 3 separate administrations. 23 Plasma-derived FVII and PCC can also be used with the same limitations as in acute bleeding. Dose and frequency can be adapted according to the clinical follow-up of the patients, 12 but evidence-based publications and guidelines are still lacking.
There is at present no guidelines for prophylaxis management regarding how and with which dose and frequency should patient undergo prophylactic schedules. A consensus to improve therapeutic schedules is needed.
Conclusion
Factor VII deficiency, whether congenital or acquired, is a rare multifaceted defect with heterogeneous laboratory features, genetic defects, and clinical manifestations. Plasma level of FVII:C determination is the cornerstone of FVII defect-specific diagnosis.
Although FVII plasma determination is required for patient’s characterization, one should keep in mind that FVII plasma level does not correlate well with the hemorrhagic diathesis. This makes both prophylaxis and bleeding management difficult for the clinician.
To conclude, FVII deficiency remains a challenge for both the clinical laboratory and the physician, and due to the lack of actual guidelines for FVII deficiency diagnosis and management, both sides should be concerned and involved jointly into optimization of patient’s care.
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are Diagnostica Stago employees.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.