
Abstract
Fourteen patients with congenital factor VII (FVII) deficiency were reported to have had pulmonary embolism. All patients were type 2 defects with variably low activity but normal or near-normal antigen. Concomitant deep vein thrombosis was present in 7 instances. The majority of patients had no or only a mild bleeding tendency. Associated prothrombotic risk factors were present in 11 patients (old age, surgery, substitution therapy with prothrombin complex, plasma-derived or activated FVII concentrates). Pulmonary embolism was usually moderate or severe. In 2 cases, it was fatal. Only 4 patients were studied by means of molecular biology techniques. The Arg304Gln mutation was found in 5 of the 8 alleles. Heparin and Coumadin together with adequate substitution therapy were carried out in 5 patients with satisfactory results. The FVII deficiency does not grant a sure protection from venous thromboembolism.
Introduction
Thrombotic events are rare in congenital bleeding disorders. Due to their high prevalence, patients with hemophilia A or B and von Willebrand disease are usually involved.1–3 Among the rare coagulation disorders, factor VII (FVII) deficiency has been reported to be more frequently associated with thrombosis.4,5 Around 50 patients with FVII deficiency have been reported to be associated with venous (more common) or arterial thrombosis (less common).5,6
A few recent articles have dealt with this peculiar association. The overall conclusion of these studies are consistent with the belief that the thrombotic manifestations are due to associated risk factors and that the clotting defect is unable to neutralize the prothrombotic impact of the associated risk factors (trauma, bed immobilization, substitution therapy, specially with prothrombin complex concentrates [PCCs] for venous thrombosis and dyslipidemia, smoking, diabetes, and hypertension for arterial thrombosis). 6
The existence of a special form of FVII deficiency prone to thrombosis was suspected but never confirmed. Several mutations have been reported in these patients.4,7,8 No article has ever dealt in a systematic pattern with pulmonary embolism (PE) in FVII deficiency. The purpose of the present study is to analyze all reported patients of FVII deficiency who manifested PE with or without concomitant deep venous thrombosis.
Patients and Methods
Personal files pertaining to the years 1968 to 2002 and 275 reports on congenital FVII deficiency have been analyzed. Sixty articles for a total of 109 cases had already been studied 9 by us in 1972. The remaining articles were analyzed after that date. Original articles were obtained through the help of the Pinali Library of our University and the courtesy of Data Med, Sorengo, Switzerland. Cross-examination of the references of the single articles was carried out in order to eliminate potential omissions. The criteria for inclusion were FVII deficiency of less than 20% of normal, a compatible hereditary pattern, and clinical and objective demonstration of the presence of PE. The objective methods for inclusions had to be pulmonary ventilatory/perfusory scintigraphy, spiral computerized axial tomography, or arteriography. The severity of the PE was graded as massive, severe, moderate, and mild according to the information supplied. Associated prothrombotic risk factors and the severity of bleeding tendency were recorded on the basis of the data presented by the authors of the single articles. The bleeding tendency was graded as severe, moderate, or mild according to the information present in each article. All patients with higher levels of FVII activity were excluded. Molecular biology studies were not an absolute requisite for inclusion. However, the mutations seen in most recent cases were recorded.
Results
At least 14 patients with FVII deficiency and proven PE were found.4,7,10–20 The main features of these patients are gathered in Table 1 . Age varied between 21 and 79 (mean 52.4). Six patients were male and 7 female. In one instance, gender was not specified. Level of FVII activity as assayed by a rabbit brain reagent was carried out in 10 cases, which varied between 1% and 15% of normal (mean 6.4). Activity of FVII using human placenta or a human recombinant preparation was carried out in only 4 patients and varied between 9 and 31 (mean 16.2). Activity of FVII using an ox brain preparation was carried out only in one instance and found to be 100% of normal. In 1 case, level of FVII was unknown, but the patient had congenital FVII deficiency. 20
Reported Cases of PE With or Without Concomitant DVT in Patients With FVII Deficiency
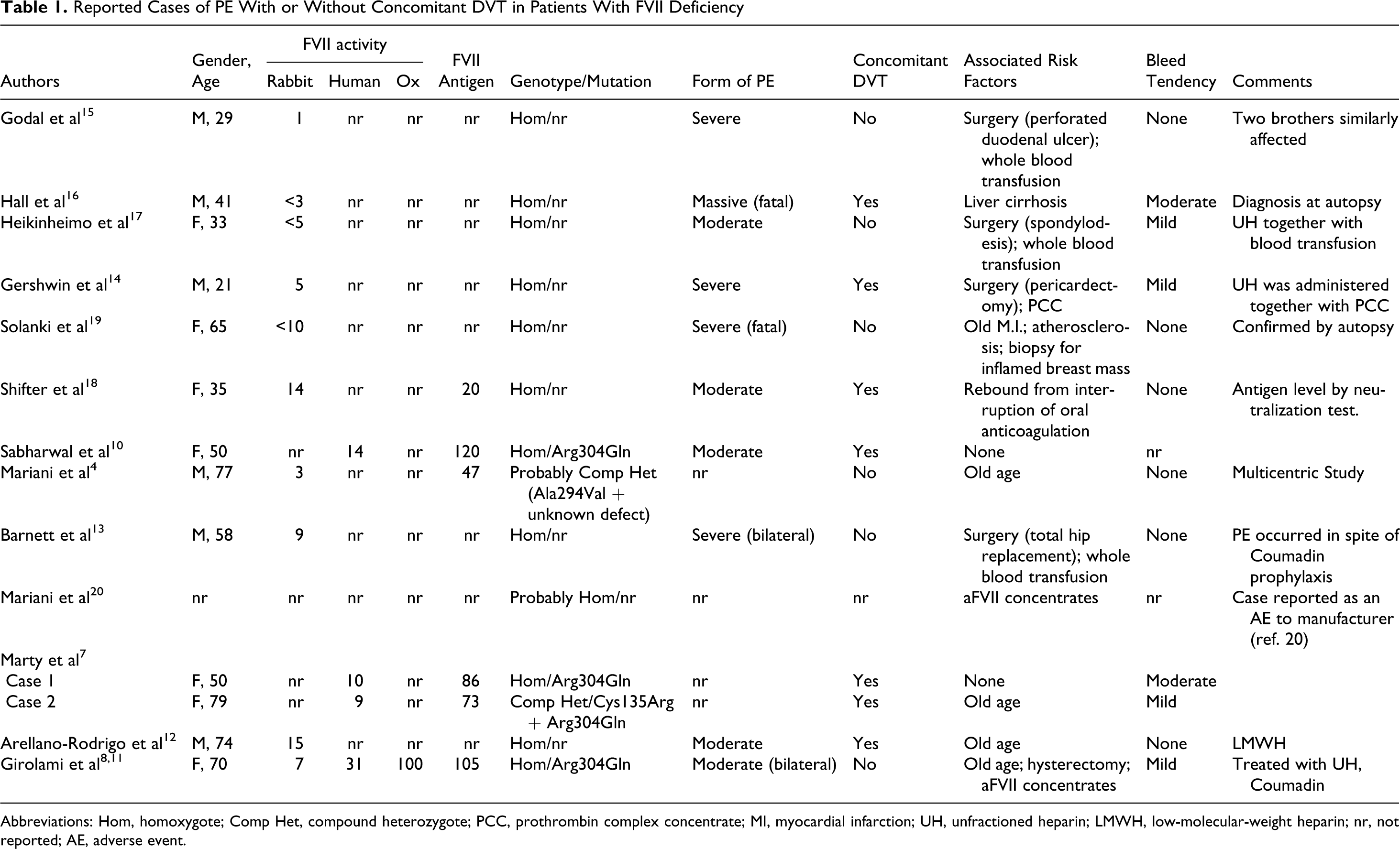
Abbreviations: Hom, homoxygote; Comp Het, compound heterozygote; PCC, prothrombin complex concentrate; MI, myocardial infarction; UH, unfractioned heparin; LMWH, low-molecular-weight heparin; nr, not reported; AE, adverse event.
The FVII antigen was evaluated only in 6 patients, and it varied between 20% and 105% of normal (mean 75%). Concomitant deep vein thrombosis (DVT) was present in 7 cases. Associated prothrombotic risk factors were present in 10 patients (old age in 4; surgery in 5; substitution therapy with whole blood, PCC, or activated FVII (aFVII) concentrates in 6; myocardial infarction or liver cirrhosis were present in 2 additional cases. Some patients had more than 1 prothrombotic risk factors. The role of associated congenital prothrombotic conditions could not be evaluated since a screening in this sense was carried out only in a few patients.11,12 There were 2 fatalities with diagnosis confirmed at autopsy. Bleeding tendency was absent in 6 patients, whereas it was mild in 4 and moderate in 2. In 2 instances, the bleeding status was not specified.
Patients were homozygotes or probably homozygous in 12 cases, whereas 2 patients were compound heterozygotes. No heterozygote was found. Molecular biology studies were reported in only 4 instances. Two patients had a homozygous Arg304Gln mutation (FVII Padua), whereas the other 2 patients were compound heterozygotes (Ala294Val + unknown defect and Cys135Arg + Arg304Gln). Altogether, the Arg304Gln mutation was present in 5 of the 8 alleles. Treatment was carried out in 5 patients and consisted of low-molecular-weight heparin or unfractioned heparin plus coumarin together with substitution therapy.
Discussion
The FVII deficiency is not known to be protective from venous thrombosis.4,6,7 The fact that in some patients, PE occurred without clinical evidence of DVT or other venous thrombosis indicates that the defect does not protect from PE.
Most of the cases presented with associated risk factors, mainly surgery, bed immobilization, and substitution therapy, with PCCs. In 2 patients, embolism was associated with aFVII concentrates (Table 1). Surgery-related cases were more frequent than for DVT. This observation indicates the need for prudent use of substitution therapy in bed-confined patients. Both PCC and aFVII preparations are implicated. The thrombogenic potential of some of these preparations even in normal individuals is well known..21,22 This concerns more PPC and aFVII than plasma-derived concentrates. However, the limited use of the latter may be a bias. It is interesting to note that in a few patients, PE occurred even after whole blood substitution therapy given for surgical procedures.15,17 This occurrence has never been reported for DVT and this remains unexplained. An increased blood viscosity due to the administration of erythrocytes could have played a role. 17
The thrombogenicity of recombinant aFVIII concentrate has received great attention in recent years. Thrombotic events occurred mainly in off-label use. 23 However, several thrombotic events were reported even in hemophilia patients with inhibitors. 21 As far as rare coagulation disorders are concerned, aFVII concentrates associated with thrombotic events have been reported also in hypofibrinogenemia and FXI deficiency.24,25
At least 6 of these patients contained cross-reacting material (CRM) in their plasma, namely they were type 2 defects. Unfortunately, the FVII antigen status of the remaining 8 patients is unknown. Only a few type I FVII-deficient patients were reported to have manifested thrombotic events. 6 This is consistent with the hypothesis that the CRM is likely to play a role.
Unfortunately, only a few of these cases have been studied by means of molecular biology techniques. The Arg304Gln is the most frequently encountered mutation but, due to the limited number of patients studied, no conclusions can be drawn. 8
There are several considerations that can be obtained from the present study, namely (1) FVII deficiency does not protect from PE; (2) PE in these patients evolves as in normal individuals; (3) once thrombosis is diagnosed, these patients should be treated as normal patients, taking into account the basal prolongation of prothrombin time and provided the defect is corrected by the use of adequate amounts of FVII concentrates. As a consequence, patients receiving FVII concentrates for therapy during surgical procedures or even for prophylaxis should be carefully monitored for the presence of signs and symptoms of PE.
Finally, it is now clear that some type II defects, for example the Arg304Gln (FVII Padua) mutation, show only a mild bleeding tendency or are asymptomatic even during surgical procedures.26,27 Therefore, these patients usually need little substitution therapy, if any. Among the 5 patients who were studied by molecular biology techniques, 2 were patients with homozygous Arg304Gln mutation (FVII Padua). This observation once again underscores the importance of a complete characterization of the clotting defect before submitting the patients to invasive and surgical procedures. A simple diagnosis of FVII deficiency may not be sufficient.26,28,29 The defect has to be characterized using at least 3 thromboplastins, namely rabbit brain, human placenta or human recombinant, and ox brain.30–32 A normal result using ox brain thromboplastin is typical of the frequently encountered Arg304Gln (FVII Padua) mutation and of the rare Arg304Trp (FVII Nagoya). 33 A normal FVII level obtained with ox brain thromboplastin suggests the presence of normal CRM.6,26
Patients with these mutations are paucisymptomatic or asymptomatic. It is worth underscoring the sharp difference in the low protection from thrombosis exercised by FVII deficiency with the absolute one manifested by FII and/or FX deficiency. 34
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: supported in part by the “Associazione Emofilia ed Altre Coagulopatie delle Tre Venezie”.