
Abstract
Venous thromboembolism (VTE) during chemotherapy is common, with 7% mortality in metastatic breast cancer (MBC). In a prospective cohort study of patients with breast cancer, we investigated whether vascular endothelial cell activation (VECA), and whether apoptosis, is the cause of chemotherapy-induced VTE.
Introduction
Venous thromboembolism (VTE) following breast cancer chemotherapy is common. In early breast cancer (EBC), VTE occurs in 1% to 10% of patients receiving chemotherapy, 1 –4 with a mortality of 0.2% to 0.5%. 1,5 The VTE rates increase to approximately 18% in metastatic breast cancer (MBC), 6 with a mortality of 7%. 6 Two possible suggestions for pathogenesis are (1) vessel wall injury as a result of the direct toxic effects of chemotherapy on endothelial cells 7 –15 and (2) apoptosis releasing procoagulant exosomes or microparticles. 16,17
The endothelial effects of intravenous chemotherapy are demonstrated clinically by the difficulty in maintaining venous access in patients. This is due to thrombophlebitis secondary to high chemotherapy concentration in peripheral venous blood proximal to the infusion site. Biochemically, chemotherapy-induced vascular endothelial cell activation (VECA) is demonstrated by increased circulating endothelial cells and plasma von Willebrand factor, markers of endothelial cell activation. 14,18 –23 Despite evidence to support chemotherapy-induced VECA, such endothelial cell activation has yet to be shown to relate to clinical VTE.
An alternate theory for chemotherapy-induced thrombosis is tumor cell apoptosis, in response to effective chemotherapy, causing prothrombotic microparticle release, 16,17 increased tissue factor (the initiating protein of the extrinsic clotting cascade) activity, and thrombin generation. 24
We aimed to determine whether chemotherapy-induced VECA or chemotherapy-induced apoptosis is the cause of VTE. In post hoc analysis of a previously reported cohort of patients with breast cancer, 25 we compared the endothelial response to chemotherapy in VTE+ compared to VTE− patients. In addition, in the metastatic subgroup of this cohort, we determined the hypercoagulable response to chemotherapy in patients with ongoing progression, despite chemotherapy (presumed low apoptosis) compared to response to chemotherapy (presumed high apoptosis).
Methods
In a prospective cohort study of patients commencing chemotherapy for breast cancer, VECA was compared in VTE+ and VTE−, and hypercoagulability was compared in patients responsive versus not responsive to chemotherapy.
Patients With Breast Cancer Commencing Chemotherapy
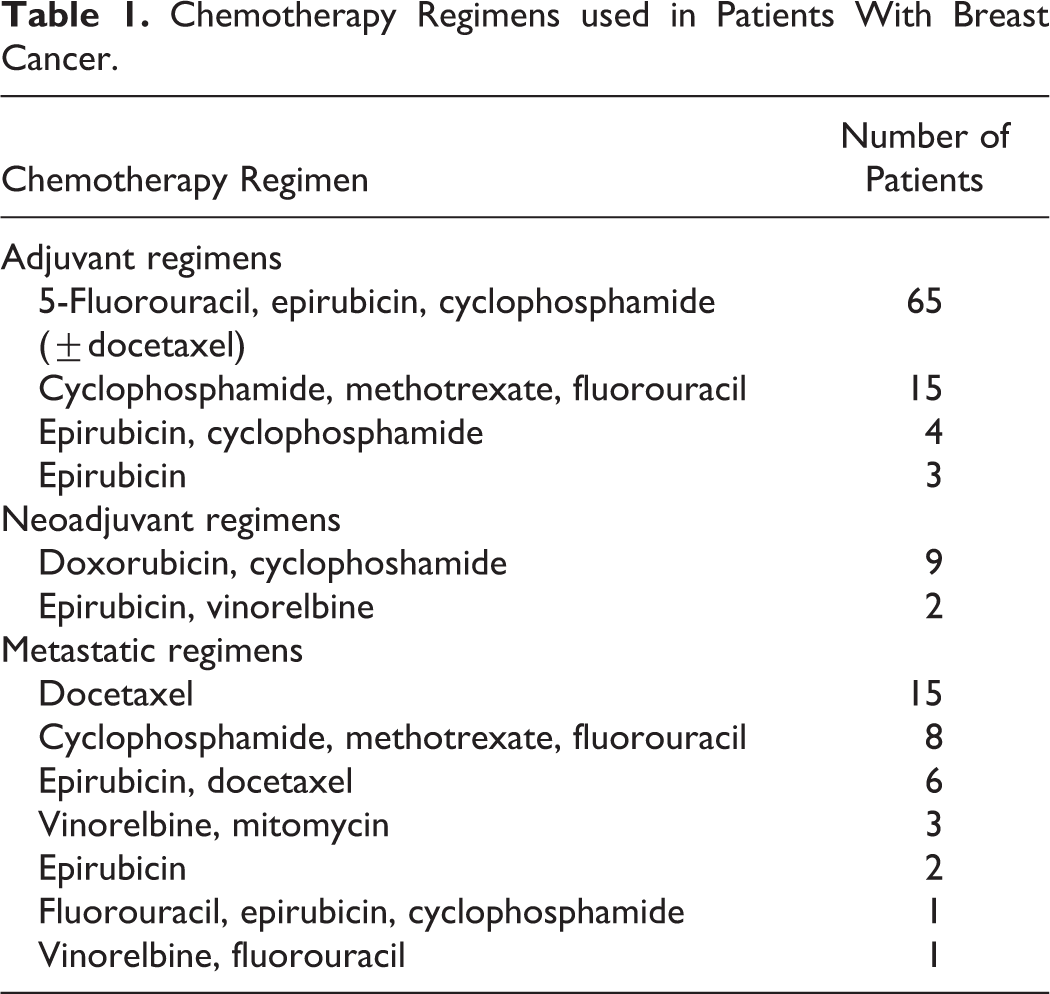
Consecutive women commencing adjuvant, neoadjuvant, or palliative chemotherapy for breast cancer at Christie Hospital NHS Trust were approached. Over a 2-year period, 134 women (median age 52 [range, 31-78] years) were recruited. Of these, 87 were patients with EBC commencing adjuvant chemotherapy following curative surgery, 11 commencing neoadjuvant chemotherapy for large or inflammatory breast cancers, and 36 commencing palliative chemotherapy for radiographically proven metastatic breast disease (Table 1). Patients were excluded if they were on anticoagulants, had a past history of VTE, had undergone previous chemotherapy within 3 months, were on hormone therapy, had implanted vascular access devices, or a World Health Organization performance status below 3. 26
Chemotherapy Regimens used in Patients With Breast Cancer.
Protocol
Serum markers of endothelial cell activation (vascular cell adhesion molecule 1 [VCAM-1] and E-selectin [E-sel]) and
Blood Sampling and Analytical Methods
Atraumatic venous blood samples, taken from the antecubital vein, were stored in citrate (plasma) or allowed to clot at room temperature (serum), separated, and stored within 2 hours. Samples were centrifuged (2500g) for 20 minutes at 4°C and the serum or plasma removed. Samples were then divided into 0.3-mL aliquots and stored at −80°C until analysis.
Serum VCAM-1 and E-sel were analyzed using enzyme-linked immunosorbent assays (ELISAs) by R&D Systems (Oxon, United Kingdom) with a sensitivity of 2 ng/mL and less than 0.1 ng/mL, respectively. Plasma
Ethical Approval
The study was approved by the South Manchester Local Research Ethics Committee, and all patients gave written informed consent.
Statistical Methods and Sample Size
Data on VCAM-1, E-sel, and
The sample size calculation was based on estimated rates of VTE in patients with MBC receiving chemotherapy compared to patients with EBC receiving chemotherapy, using a chi-square test with Yates correction. 4,6 This study was originally powered to detect a difference in rate of VTE between patients with MBC and EBC, with 120 patients in each group providing 80% power to detect a difference (5% significance) between the expected frequency of VTE of 17% with chemotherapy for advanced cancer and 5% for early carcinoma. The study was closed before recruitment targets were reached.
Results
Of 134 patients with breast cancer commencing chemotherapy, 10 (7.5%) patients were VTE+ of which 6 (60%) were symptomatic (Table 2).
Demographics and VTE Data on Patients Developing VTE.
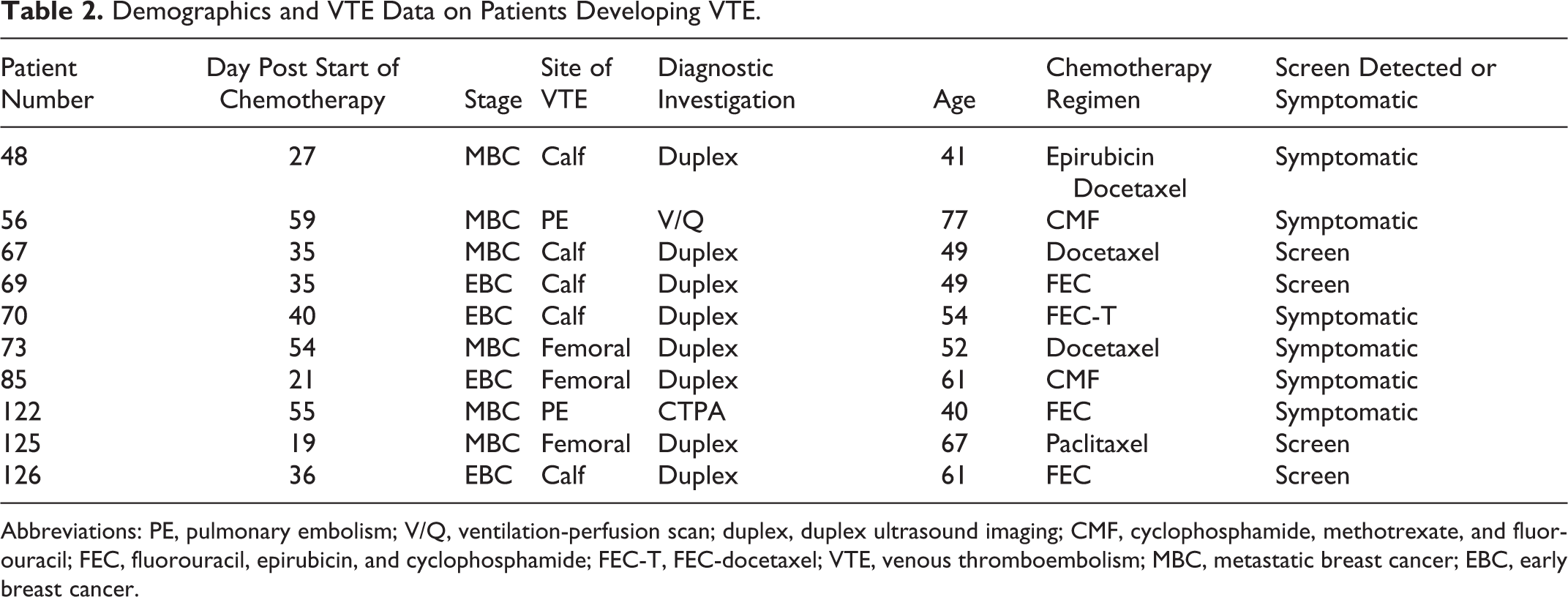
Abbreviations: PE, pulmonary embolism; V/Q, ventilation-perfusion scan; duplex, duplex ultrasound imaging; CMF, cyclophosphamide, methotrexate, and fluorouracil; FEC, fluorouracil, epirubicin, and cyclophosphamide; FEC-T, FEC-docetaxel; VTE, venous thromboembolism; MBC, metastatic breast cancer; EBC, early breast cancer.
No patients were lost to follow-up. Of the 36, 6 (17%) patients with MBC and of the 87, 4 (4.6%) patients with EBC were VTE+. The increased rate of VTE in MBC compared to EBC approached significance (P = .06). None of the 11 patients receiving neoadjuvant chemotherapy developed VTE. At baseline,
The E-sel, VCAM-1, and
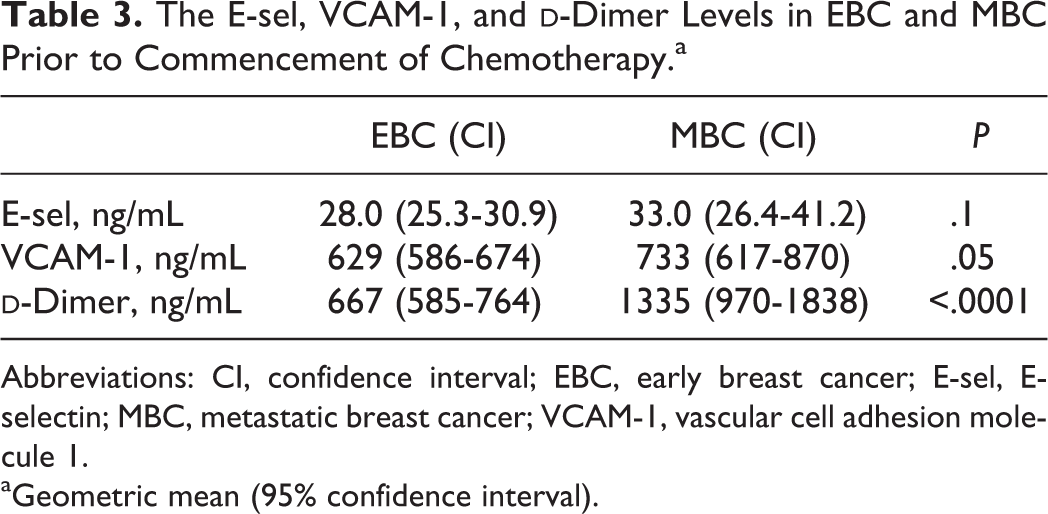
Abbreviations: CI, confidence interval; EBC, early breast cancer; E-sel, E-selectin; MBC, metastatic breast cancer; VCAM-1, vascular cell adhesion molecule 1.
aGeometric mean (95% confidence interval).
Baseline Hypercoagulability and Endothelial activation
Baseline
The E-sel, VCAM-1, and
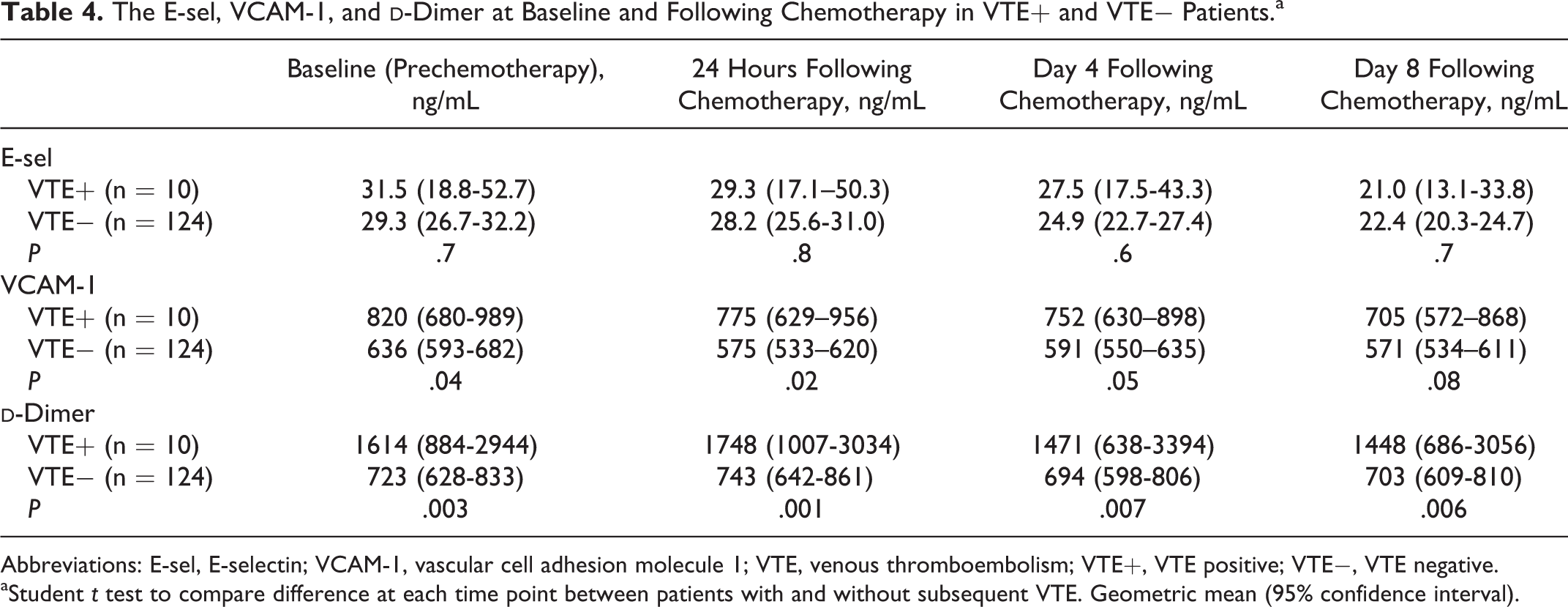
Abbreviations: E-sel, E-selectin; VCAM-1, vascular cell adhesion molecule 1; VTE, venous thromboembolism; VTE+, VTE positive; VTE−, VTE negative.
aStudent t test to compare difference at each time point between patients with and without subsequent VTE. Geometric mean (95% confidence interval).
Chemotherapy-Induced VECA in VTE+ Compared to VTE− Patients
Analyzing all patients, irrespective of subsequent development of VTE, both E-sel and VCAM-1 decreased following chemotherapy (repeated measures analysis, P < .001), suggesting an endothelial response to chemotherapy. E-sel and VCAM-1 did not correlate with markers of the acute-phase response such as CRP, platelet count, and fibrinogen. However, this decrease was similar in VTE+ and VTE− patients (Figures 1 and 2), implying no difference in VECA between VTE+ and VTE−. Separate analysis comparing symptomatic VTE+ and VTE− also revealed no difference in VECA. Surprisingly,
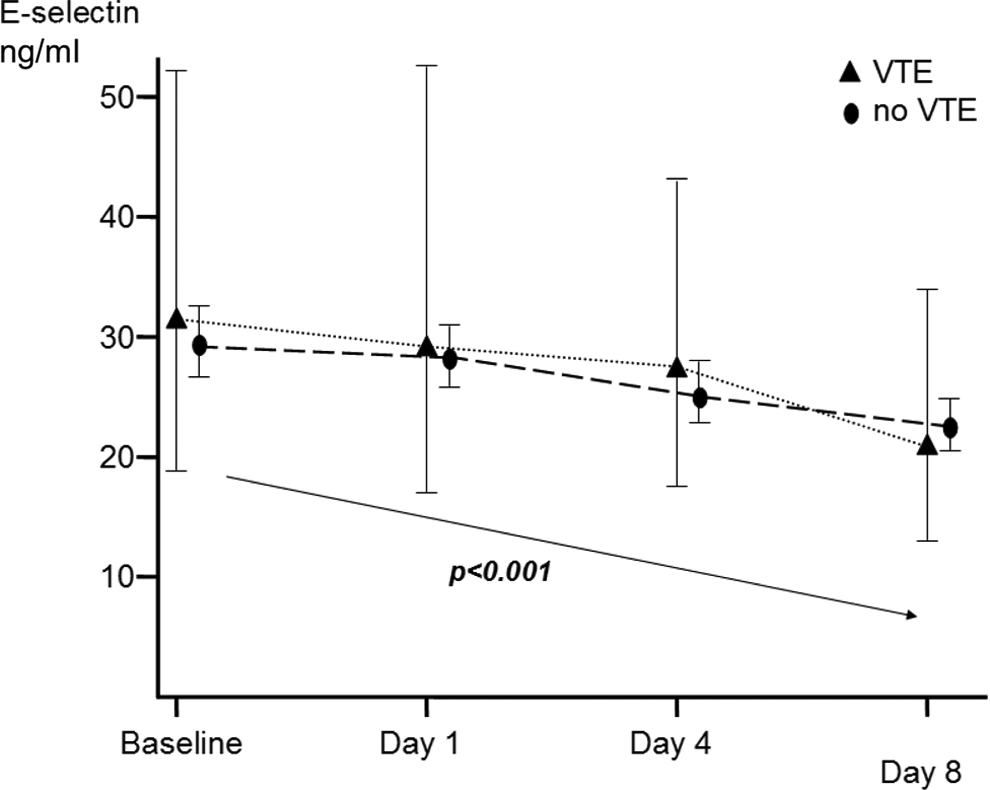
Change in serum E-selectin (E-sel) in response to chemotherapy in venous thromboembolism-positive (VTE+) and venous thromboembolism-negative (VTE−) patients. Geometric mean. Repeated measures analysis with Greenhouse-Geiser correction.
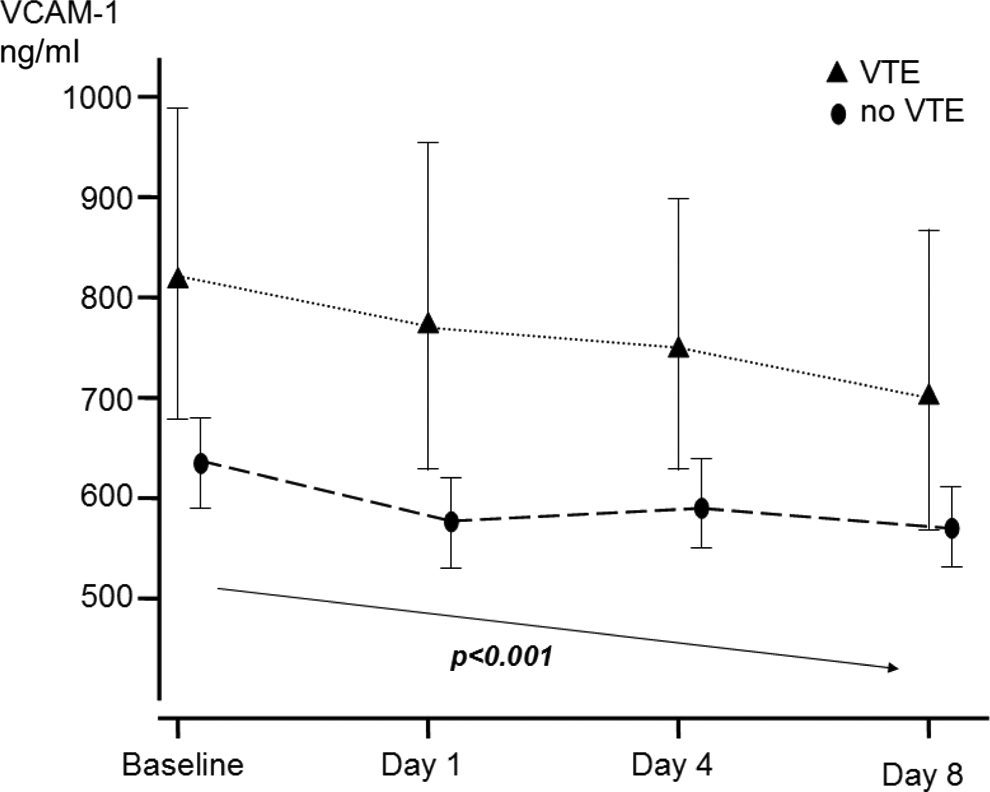
Change in serum vascular cell adhesion molecule 1 (VCAM-1) in response to chemotherapy in venous thromboembolism-positive (VTE+) and venous thromboembolism-negative (VTE−) patients. Geometric mean. Repeated measures analysis with Greenhouse-Geiser correction.
Changes in E-sel did not correlate with
Hypercoagulable Response to Chemotherapy in MBC With early Progression Compared to Patients Responding to Chemotherapy
In subgroup analysis of the 36 patients with MBC, 15 had cancer progression at 3 months. In the 8 days following commencement of chemotherapy,
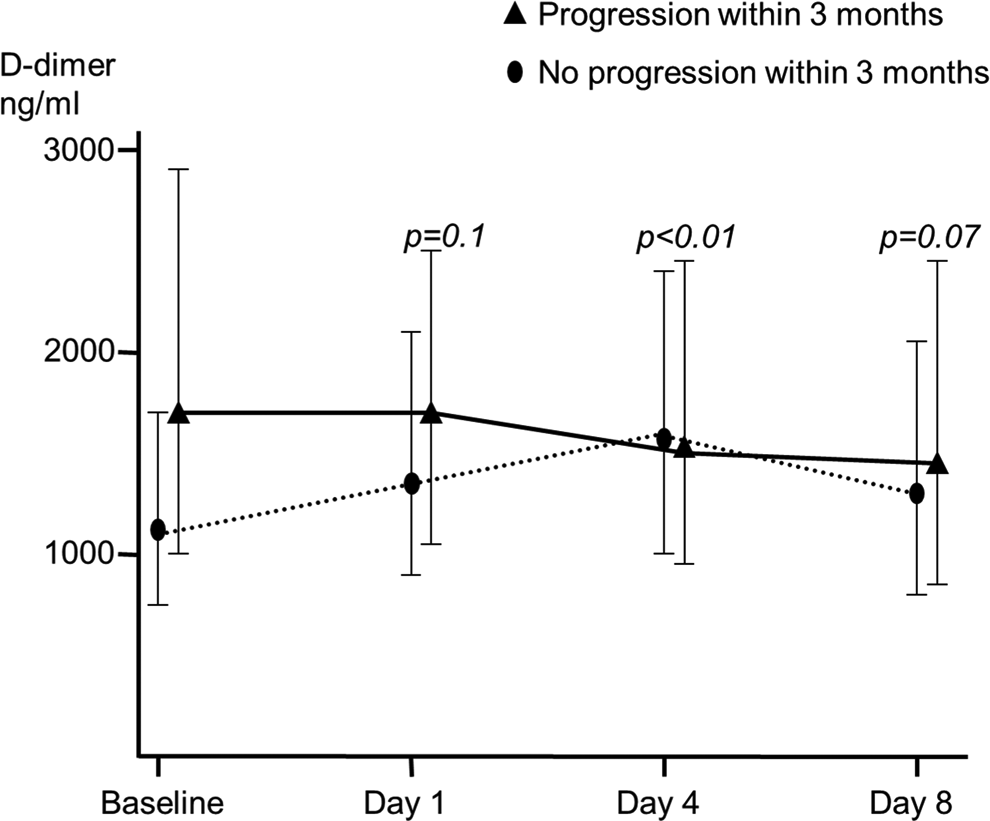
Change in plasma
Discussion
The chemotherapy-induced VTE rate of 4.6% in EBC and 17% in MBC reported in this study supports previously quoted figures.
1
–3,6,25
The risk of chemotherapy-induced VTE is underrecognized,
27
and thromboprophylaxis during chemotherapy is rarely used.
27
Irrespective of the cause of VTE, mortality from pulmonary embolism (PE) is 5% to 17%.
28
Development of VTE has an important impact on the mortality and morbidity of patients with cancer. Up to 60% of patients dying of cancer-associated VTE have localized cancer or limited metastatic disease, which would have allowed for reasonable survival in the absence of fatal PE.
29
In addition, the clinical course of VTE is complicated in up to 30% of patients by development of postthrombotic sequelae, such as pain, edema, hyperpigmentation, and ultimately venous ulceration.
30
Following development of VTE, there is a risk of VTE recurrence of 25% at 5 years, with this risk almost doubled in patients with cancer.
30
We have previously reported that a normal
Bolus chemotherapy administration causes the vascular endothelium to be temporarily exposed to high concentrations of circulating toxic drugs, prior to peripheral tissue uptake. In addition, many chemotherapies have specific endothelial cytotoxic effects. At clinically relevant doses, bleomycin, vincristine, and adriamycin cause endothelial cell retraction with exposure of subendothelial matrix in vascular endothelial cell monolayers. 31 Paclitaxel, cyclophosphamide, methotrexate, and fluorouracil are all cytotoxic to endothelial cells. 32 –35 Doxorubicin and cisplatin induce endothelial cell apoptosis, with the later causing release of endothelial microparticles. 17,36 Cultured human endothelial cells incubated with the plasma of patients with breast cancer receiving monthly adjuvant cyclophosphamide, epirubicin and 5-fluorouracil chemotherapy demonstrated increased endothelial cell reactivity 13 providing further evidence that chemotherapy induces endothelial cell activation. In support of this, we have shown a marked endothelial response to chemotherapy, with both E-sel and VCAM-1. The E-sel is a recognized marker of endothelial cell activation 37 that has been shown, in mouse models, to be important for intravascular thrombus formation. 38 Levels of E-sel are increased in patients with myeloproliferative disease and venous thrombosis compared to patients with myeloproliferative disease without thrombosis. 39 The VCAM-1 is an endothelial adhesion protein. Expression of VCAM-1 is induced on endothelial cells by inflammation, where it acts as an adhesive receptor to facilitate adhesion, migration, and transmigration of circulating leukocytes into damaged vascular tissues. Circulating levels are increased in vasculitis. 40 There is a close relationship between inflammation and VECA; however, we found no correlation between VECA and markers of the acute-phase response such as platelet count, fibrinogen, or CRP in our cohort of patients, highlighting that E-sel and VCAM are not simply surrogate markers of inflammation.
The E-sel, VCAM-1, and
The lack of baseline duplex ultrasound imaging is a potential weakness of this study; however, all patients were asymptomatic at recruitment. More importantly, chemotherapy-induced VTE in breast cancer is well documented, with baseline VTE rates in breast cancer one of the lowest cancers. A recent UK epidemiological study of 13 202 patients with breast cancer has highlighted that VTE in breast cancer is primarily associated with chemotherapy, with a greater than 10-fold increased risk of VTE following chemotherapy. 43
It is noteworthy that in our study both plasma E-sel and VCAM-1 decreased over time rather than increased. This may represent increased uptake of the circulating molecules by endothelial cells to allow increased expression of E-sel and VCAM-1 by activated endothelial cells, a marker of endothelial cell activation. 44 A similar response was seen in a study of patients undergoing hip surgery, with reducing E-sel levels, and stable VCAM-1 in response to the endothelial cell stimulus of surgery. 45,46 This hypothesis of particular polymorphisms of E-sel anchoring to the endothelial surface, thereby limiting the shedding of this molecule into the bloodstream is supported by the inverse relationship between E-sel and prognosis in end-stage renal disease. 47
However, this study was designed to investigate whether such chemotherapy-induced endothelial cell activation correlated with development of VTE. Many authors have assumed this causal relationship. 7 –15 In vitro, Swystun et al demonstrated that exposure of plasma to either doxorubicin- or epirubicin-treated endothelial cells resulted in an increase in plasma thrombin generation. 48 However, for the first time, in this study we have demonstrated that there is no increased endothelial response in VTE+ compared to VTE− patients.
Although we cannot exclude the possibility that VECA plays a minor role in the multifactorial complication of chemotherapy-induced VTE, this study shows that endothelial cell activation is not a primary mechanism for causing VTE.
As patients with EBC are to some extent representing patients who are “cancer free” yet receiving chemotherapy, it may be reasonable to assume that VTE in this group is truly chemotherapy induced. The increased rate of VTE in MBC, compared to EBC, in our study implies that VTE is not simply a “chemotherapy” effect but may also be a “tumor response to chemotherapy” effect. The increased VTE rate in MBC supports the hypothesis of chemotherapy causing procoagulant release from the tumor (a factor that is missing in the EBC group, if it is presumed that patients with EBC are largely tumor free). This hypothesis is further strengthened by the chemotherapy-responsive patients having the greatest hypercoagulable response to chemotherapy. Patients with MBC responding to chemotherapy will have increased early tumor death or apoptosis, potentially enhancing the release of tumor-derived procoagulant exosomes or microparticles into the systemic circulation. This finding has important clinical implications, as it suggests that it is the patients who are responding to chemotherapy that may be succumbing to VTE. This increases the importance of thromboprophylaxis in this subgroup, as they will have a relatively promising cancer prognosis, compared to nonresponding patients with MBC. It is however acknowledged that patient numbers are limited in this subgroup and so this is hypothesis-generating data.
Conclusion
This study provides evidence that chemotherapy-induced VTE is not primarily due to endothelial cell activation. It further provides evidence that chemotherapy-induced apoptosis may initiate VTE. This may allow identification of patients at increased risk of VTE and allow more targeted thromboprophylaxis.
Footnotes
Authors’ Note
Presented at International Conference of Thrombosis and Haemostasis Issues in Cancer, Bergamo, Italy, April 2010.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: During the course of this study, CCK was in receipt of a Royal College of Surgeons of England research fellowship.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by Cancer Research UK (grant number GD253EYS) and the Royal College of Surgeons of England.