
Abstract
The aim of this study was to detect the genetic alterations in the Factor 8 gene in 26 patients from Western Algeria. We detected the presence of “intron 22 inversion” with long-range polymerase chain reaction (PCR). Negative patients for this inversion were analyzed for “intron 1 inversion” using multiplex PCR. Patients who were negative for both inversions were analyzed using a direct sequencing. Deleterious effects of novel mutations on protein were assayed with bioinformatics tools. Causing mutations were identified in 85.71% of the families, including 11 “intron 22 inversion,” 1 “intron 1 inversion,” and 6 different point mutations (2 nonsense, 1 splice site, and 3 missense mutations). Among these mutations, c.2189G > A (p.Cys711Tyr) and c.5219+1G>T are novel. This is the first study that reports spectrum of mutations in the Factor 8 gene in the Western Algerian population. Knowledge of these mutations is important for genetic counseling and medical care of affected families.
Introduction
Hemophilia A (HA; OMIM: 306700) is the most severe X-linked recessive bleeding disorder affecting approximately 1 of 5000 male births. It is caused by absence or functional abnormality of factor VIII (FVIII), a plasma glycoprotein that plays an important role in amplification phase of blood coagulation. 1 According to the residual plasma FVIII coagulant activity (FVIII: C), HA can be classified into 3 forms: severe (FVIII: C < 1%), moderate (1% < FVIII: C < 5%), and mild (5% < FVIII: C < 40%). 2
Factor 8 gene (F8) is located on the distal end of the long arm of the human X chromosome (Xq28) and consists of 26 exons, spanning 186 Kilo bases (kb) of genomic DNA. 3 F8 messenger RNA (mRNA) of approximately 9 kb encodes a precursor protein of 2351 amino acids (AAs), consisting of 19 AAs of the leader peptide followed by 2332 AAs of the mature protein. 4 This mature protein is divided into 6 distinct domains and arranged in the order: A1-A2-B-A3-C1-C2. 5
HA is a genetically heterogeneous disorder resulting from wide range of mutations that have been reported in international databases: Haemophilia A Mutation, Structure, Test, and Resource Site (HADB/HAMSTeRS), Human Gene Mutation Database (HGMD), and the CDC Hemophilia A Mutation Project (CHAMP). 6 –8 The only common gene defects in HA are intron 1 and 22 inversions, which occur in, respectively, 5% and 45% to 50% of patients with severe form. 9,10 The other mutations comprise large and small deletions, insertions, duplications, and point mutations resulting in nonsense, missense or splice site mutations, or other gene rearrangements. 11
It is well documented that mutation screening in the F8 gene is important because it contributes to detection of carrier women, prenatal testing for affected pregnancies, and even fostering newer therapeutic strategies. 12 Moreover, this analysis contributes to appropriate patient care decisions. In fact, genotype/phenotype correlation helps in prediction of the likelihood of FVIII inhibitor production in patients with HA, the most serious complication in HA treatment to date. 12
Several studies have focused on the molecular defects of F8 gene in different ethnic groups but not in Algeria, where the disease incidence was reported to 4.28 per 100 000 Algerian males, according to the World Federation of Hemophilia Report on the Annual Global Survey 2011.
This is the first study that reports spectrum of mutations in the F8 gene responsible for HA in the Western Algerian population. First, we detected the HA-causing mutations using different molecular methods. Then, we performed an analysis of deleterious effects of novel mutations detected for a better understanding of the structure–function relationship of the mutate FVIII protein. Finally, we discussed spectrum of mutations regarding the phenotype and status of inhibitor development of our patients.
Patients and Methods
Patients
A total of 26 Algerian patients with HA (21 unrelated families) aged between 4 and 46 years were recruited between 2011 and 2012 from Oran treatment center of HA in the West of Algeria. Informed written consent, according to the declaration of Helsinki, of every patient or their parents was obtained. For each patient, 2 blood samples were collected in tubes containing, respectively, citrate and EDTA for coagulation and genetic analysis.
Methods
F VIII activity and FVIII inhibitor analysis: factor VIII: C was measured using the STA-Deficient VIII reagent (Diagnostica Stago, Asnieres, France). Factor VIII inhibitor screening and titration were performed according to Bethesda/Nijmegen modification assays. 13
DNA extraction: genomic DNA was isolated from 5 mL of peripheral blood cell samples using a commercial DNA extraction kit according to the manufacturer’s instructions (Stratagene DNA Isolation Kit; Agilent, Canada).
Detection of intron 22 and intron 1 inversions: patients with severe form of HA were first screened for intron 22 inversion using a long-range polymerase chain reaction (PCR). 14 Intron 1 inversion was assessed by multiplex PCR. 10
Sequencing of F8 gene: after excluding patients having intron 22 or 1 inversions, all 26 exons including flanking intronic sequences, promoter, and 3′UTR were amplified using primers and PCR condition described in the HAMSTeRS database. 6 The amplified fragments were purified and sequenced using ABI Prism 310 Genetic Analyzer with BigDye Terminator v3.1 kit (Applied Biosystems, Carlsbad, California).
Molecular modeling: the possible functional alteration was evaluated using different bioinformatics tools exploring impact on protein and splicing. Protein effect: (1) PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), (2) Sort Intolerant from Tolerant (SIFT; http://blocks.fhcrc.org/sift/SIFT.html), and (3) GVGD (http://agvgd.iarc.fr/agvgd_input.php) were used to study the effect of missense mutations on protein structure. (4) Swiss Pdb Viewer software Version 4.0.4 (http://www.expasy.org/spdbv; Swiss Institute of Bioinformatics, Basel, Switzerland) was used to study missense mutation site in the 3.98 Å x-ray structure described by Ngo et al (Protein Data Bank ID: 3CDZ). 15 Splicing effect: Human Splicing Finder (HSF; http://www.umd.be/SSF) was used to evaluate the effect of splice site mutations.
Results
Baseline characteristics of the 26 Algerian patients with HA (21 unrelated families) are summarized in Table 1. Laboratory phenotype data and bleeding history (data not shown) allowed us to classify 19 families with severe and 2 with moderate HA.
Baseline Characteristics of the 21 Algerian Hemophilia A Families.
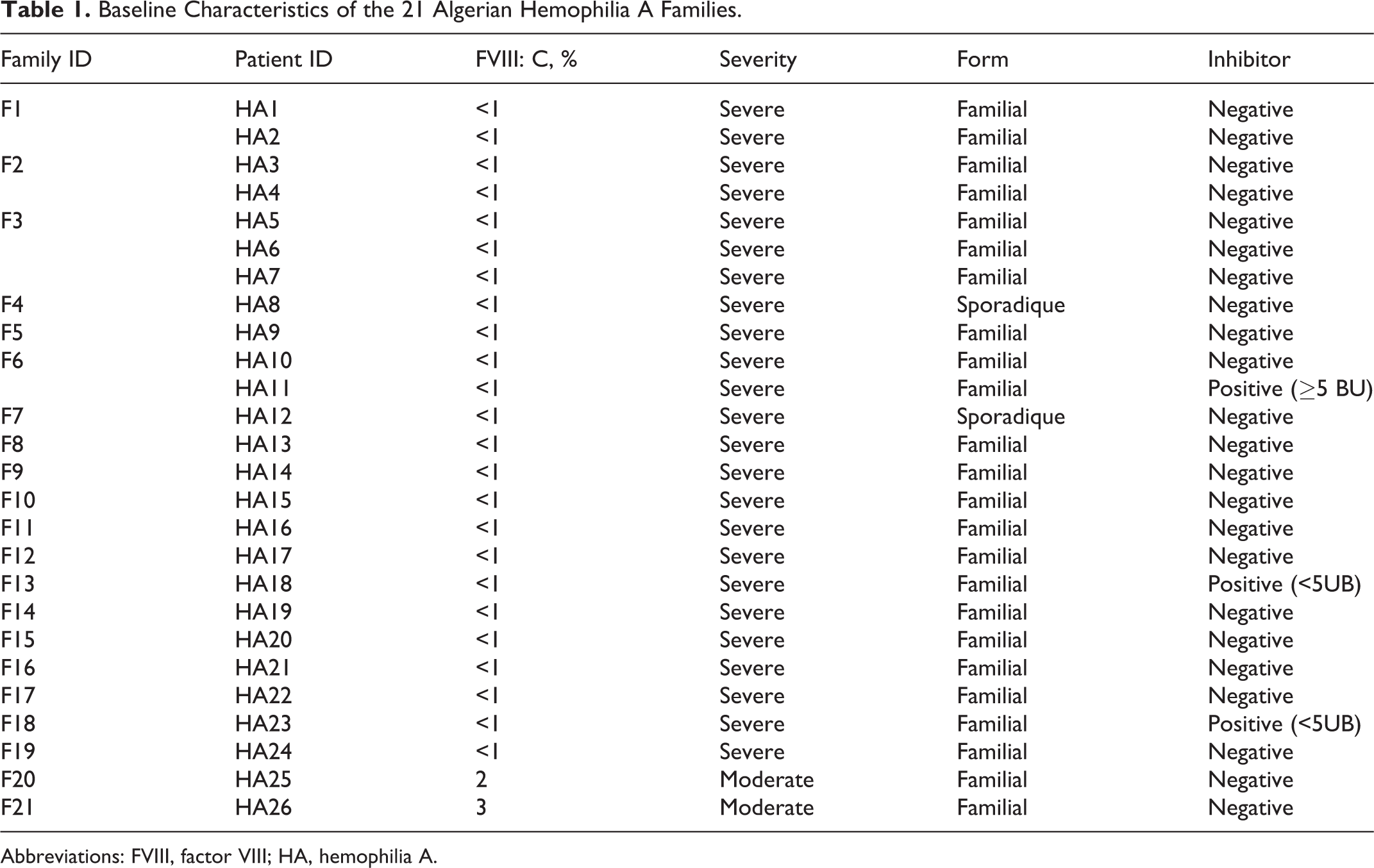
Abbreviations: FVIII, factor VIII; HA, hemophilia A.
In this study, we identified 8 different mutations including intron 22 inversion, intron 1 inversion, and 6 different point mutations. No large deletion, insertion, or other gene rearrangement were detected in our patients with HA.
Intron 22 and Intron 1 Inversions
Intron 22 inversion was detected in 11 (57.89%) among the 19 unrelated families with a severe form of HA (Table 2). Intron 1 inversion was found in only 1 patient (HA17) from intron 22 inversion negative subgroup investigated (5.26%). As intron 1 and 22 inversions are well established as causative mutations in severe form of the disease, no further investigations for other mutations in coding regions in these families were realized.
Frequencies of Intron 22 and Intron 1 Inversions.

Point Mutations
A description of the identified mutations in the remaining patients of this study is reported in Table 3. Six different point mutations (2 nonsense, 1 splice site, and 3 missense mutations) and 1 polymorphism were identified.
List of Causative Point Mutations Detected in the Algerian Patients With HA.
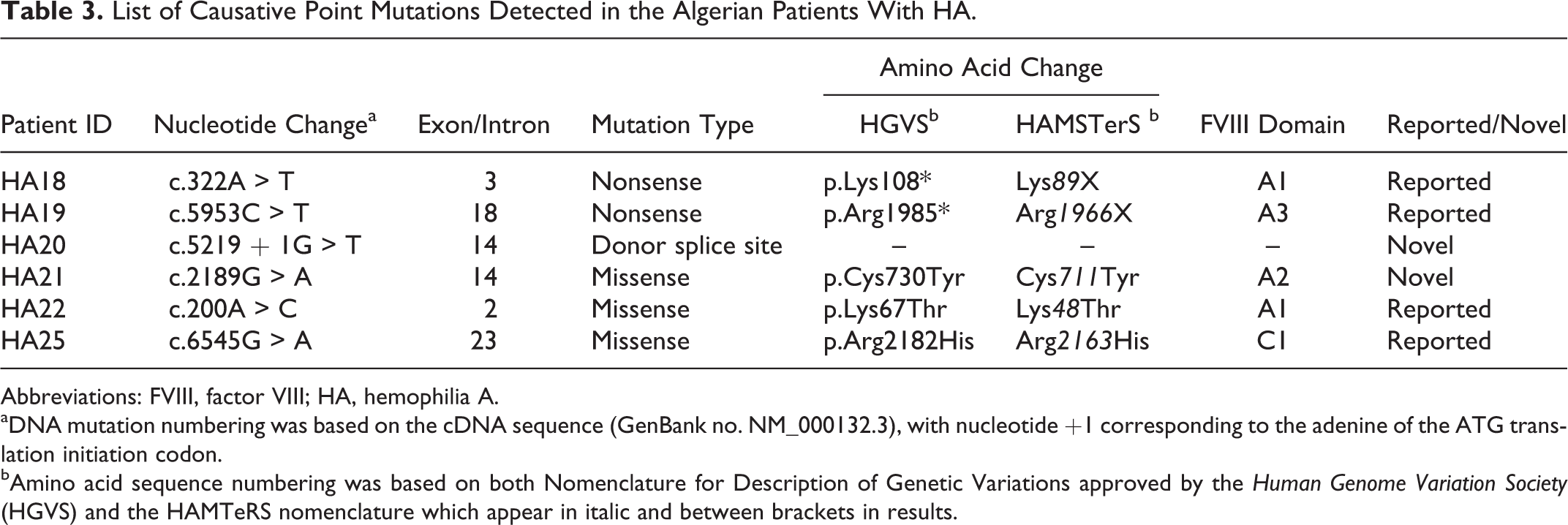
Abbreviations: FVIII, factor VIII; HA, hemophilia A.
aDNA mutation numbering was based on the cDNA sequence (GenBank no. NM_000132.3), with nucleotide +1 corresponding to the adenine of the ATG translation initiation codon.
bAmino acid sequence numbering was based on both Nomenclature for Description of Genetic Variations approved by the Human Genome Variation Society (HGVS) and the HAMTeRS nomenclature which appear in italic and between brackets in results.
Nonsense and splice site mutations: 2 nonsense mutations (c.322A > T, p.Lys108*; c.5953C>T, p.Arg1985*) were identified. These mutations are, respectively, located at position 108(89) in exon 3 and at position 1985(1966) in exon 18 of the F8 gene and result in truncated proteins considered as pathogenic (FVIII: C<1%). Both of them are already described in HAMSTeRS database.
Only 1 splice donor site mutation (c.5219 + 1G > T) was detected in patient with severe HA and consists of a transversion G > T at the first nucleotide of intron 14 (Table 3). This novel mutation was not described in any of the mutation databases or in published articles. Analysis of the possible effect of this mutation on F8 pre-mRNA splicing by HSF software predicts abolition of the normal splice donor site (score < 0.4).
Missense Mutations: 3 missense mutations were identified in 3 unrelated families, 1 with moderate HA and the remaining families with a severe form of the disease (Table 3).
The missense mutation c.2189G > A, (p.Cys730Tyr) was detected in exon 14 and consists of a G > A transition at codon TGT (cysteine) to TAT (tyrosine) at position 730 (711) of the A2 domain of the FVIII protein (Figure 1).This mutation was not described in any of the mutation databases or in published data.
Analysis of the effect of this novel mutation using in silico methods shows that the cysteine residue is conserved among mammalian species (canine, murine, and pig). Furthermore, this mutation was categorized as “Probably damaging” and “Not Tolerated” using Polyphen2 and SIFT softwares. Analysis with GVGD Align shows that the p.Cys730Tyr (c.2189G > A) was considered as “less likely interfere with function.” In addition, the effects of AAs substitution on protein structure were analyzed using Swiss Pdb Viewer software and clearly show an impact on the structure of the mutated p.Cys730Tyr (c.2189G > A) protein (Figure 2A).
Among the 4 substitutions detected, the c.200A > C (p.Lys67Thr) mutation is located in the second exon of the F8 gene. This mutation consists of a A > C transversion and causes a change in the second base of the codon AAG (lysine) to ACG (threonine) at position 67 (48) of the A1 FVIII domain (Figure 1). This mutation has been recently reported in the HAMSTeRS database. However, no functional analysis of this substitution using in silico methods was carried out. The substitute residue is conserved among mammalian species (canine, murine, and pig) and is categorized as “Probably damaging” and “Not Tolerated” using Polyphen2 and SIFT softwares. Analysis with GVGD Align shows that the p.Lys67Thr (c.200A > C) was considered as “most likely interfere with function.” The effect of this mutation on protein structure is shown in Figure 2B.
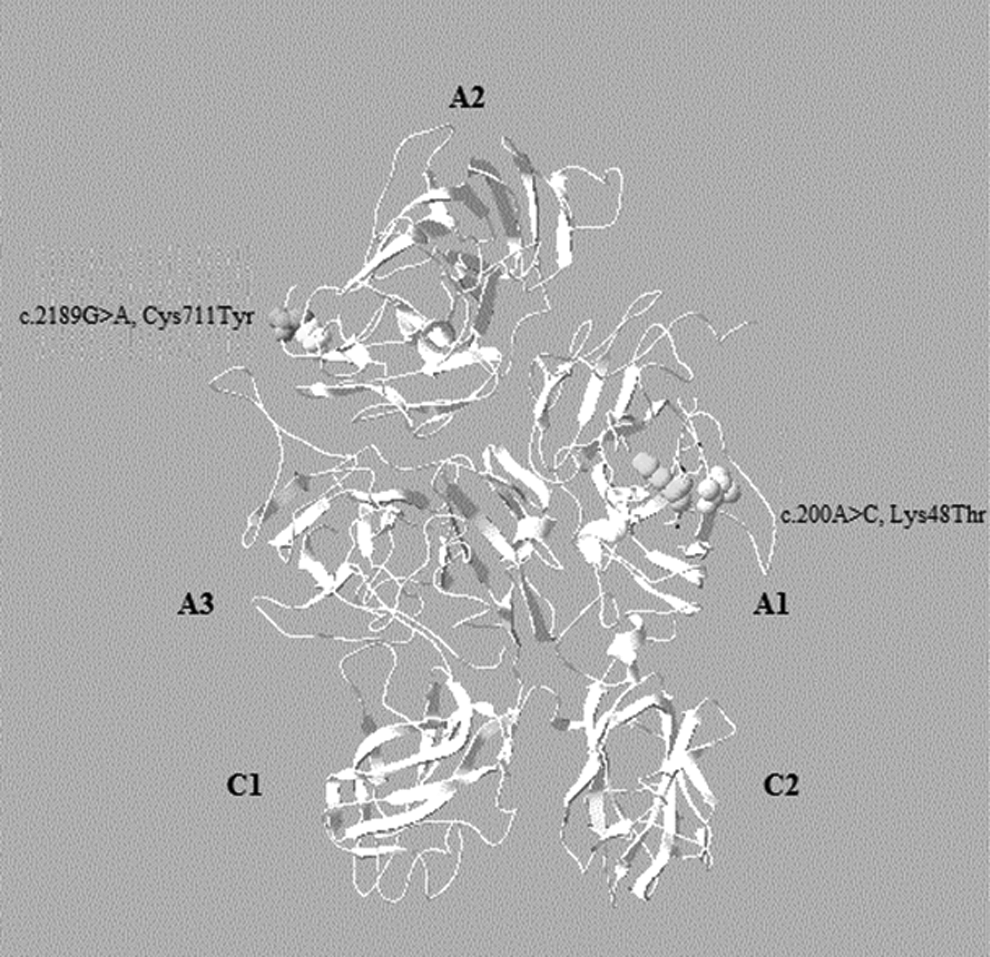
Location of the 2 missense mutations p.Lys48Thr and p.Cys711Tyr on 3-dimensional model of human FVIII protein. Ribbon diagram of human FVIII protein was produced using Swiss Pdb Viewer 4.0.4 software and Protein Data Bank entry 3CDZ. Heavy-and light-chain domains of FVIII protein are shown (A1.A2.A3.C1.C2). FVIII indicates factor VIII.
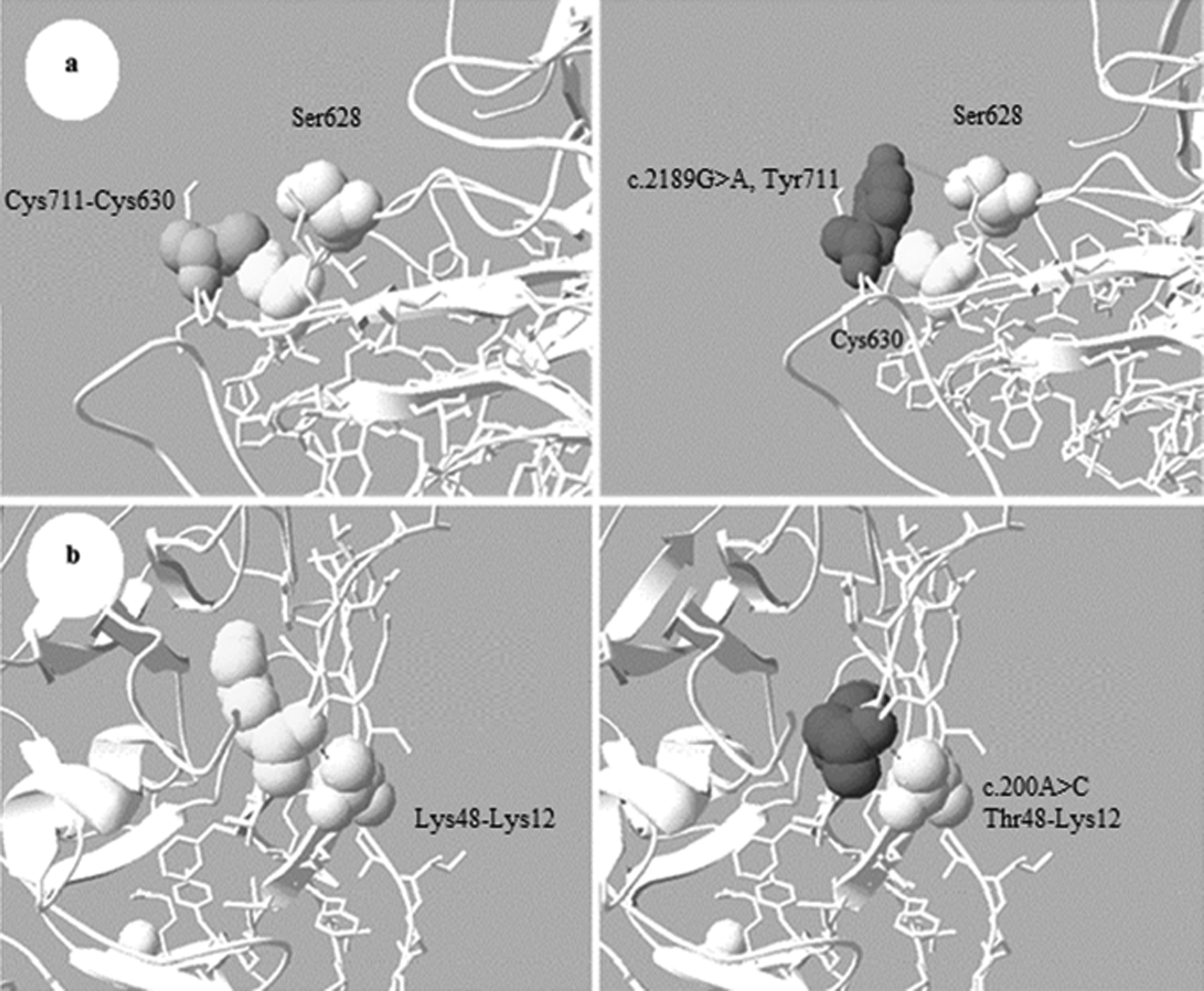
Prediction of the deleterious effects of p.Lys48Thr and p.Cys711Tyr using Swiss Pdb Viewer 4.0.4 software. A, Left: the normal interaction with a disulfide bond between cysteine 711 and cysteine 630. Right: molecular modeling of effect of the p.Cys711Tyr on protein structure. B, Left: the normal interaction between lysine at position 48 with lysine at position12. Right: molecular modeling of effect of the p.Lys48Thr on protein structure.
The reported missense mutation c.6545G > A (p.Arg2182His) was identified in 1 patient with moderate form of HA. This mutation occurs in the 23rd exon and causes a change in the second base of the codon CGT (arginine) to CAT (histidine) at position 2182 (2163) in the C2 domain. Analysis of this mutation with SIFT and Polyphen2 revealed that it was, respectively, “not tolerated” and “possibly damaging.”
Polymorphism: for HA 23 patient, the only genetic event detected was a variant in exon 14 (c.3780G > C, p.Asp1260Glu). This variant causes a change in the third base of the codon GAC (aspartic acid) to GAG (glutamic acid) at position 1260 (1241) in the B domain.
Patients With HA Without an Obvious Causative F8 Mutation
No obvious F8 mutation was found in HA23, HA24 and HA26 patient even after sequencing the coding F8 region, promoter, and 3′UTR and searching for large rearrangement with a multiplex ligation probe amplification (data not shown).
F8 Mutations and Inhibitor Development
Information about development of inhibitors was available for all patients (Table 1). Inhibitors were found in 12.5% (3 of 24) of the patients with severe form of HA: 1 with nonsense mutation (c.322A > T, p.Lys108*), 1 with the polymorphism (c.3780C > G, p.Asp1260Glu), and 1 with intron 22 inversion.
Discussion
The first aim of this study was to detect HA causative mutations in 26 patients with HA belonging to 21 unrelated families in Western Algeria. Molecular methods used for the detection of intron 22 and intron 1 inversions were easy to perform, highly reproducible, and eliminate the need for the more laborious and costly analysis of the polymorphic site. For patients negative for these 2 inversions and in accordance with the previous studies, 16,17 we used a direct sequencing technique that matches the best cost–time ratio for the detection of the causative alteration throughout the F8 gene. In 3 families, the responsible mutation is still unknown, but the total analytical sensitivity of our strategy is estimated to be 85.71% (18 of 21). It was previously reported that in approximately 2% of patients with HA, no genetic mutation could be found in the F8 even after sequencing including the promoter, the entire coding region, exon/intron boundaries, and the 3′UTR. 18,19 In these cases, the possibility that some causative mutations might be located in an unanalyzed region of F8 is still suspected. It is difficult to examine deep inside intron in detail, which leaves this relatively unanalyzed region as a strong candidate for undetected mutations. 20
Intron 22 and Intron 1 Inversions
The incidence of intron 22 inversion in our study was equal to 57.89%, which is slightly higher than that in other populations from Mexico, Venezuela, and Germany. 21 –23 The frequency of the intron 1 inversion observed in our study (5.26%) was within the range reported in the articles. This inversion was initially reported to be responsible for approximately 5% of patient with severe HA. 21 However, recent studies suggest that its overall incidence is lower than 1%. 23,24 When we compare our results with previous data from Arab countries, the frequencies of these 2 inversions are higher in our patients. 17,25 –27 In fact, intron 22 inversion frequency was reported between 23.3% and 55% in Egypt, Tunisia, and Saudi Arabia, whereas any of the intron 1 inversion was detected. These results could suggest that our patients have a specific genetic profile. Nevertheless, these data must be confirmed with a larger number of studied patients. We assume that this difference in frequencies result most likely to the relatively small number of patients studied.
Point Mutations
All patients without intron 1 and 22 inversions displayed a specific mutation that confirmed the high heterogeneity of causative F8 alteration in the Algerian population.
Nonsense and splice site mutations: the deleterious mechanisms of nonsense mutations are in general obvious given that they create premature termination codons leading to truncated F8 mRNA and FVIII protein. 28 The truncated mutant protein caused by the c.322A > T (p.Lys108*) would just contain a small part of the A1 domain, whereas truncated mutant FVIII caused by the c.5953C > T (p.Arg1985*) would lack A3, C1, and C2 domains and are both severely impaired proteins.
Concerning the splice donor site mutation, analysis with HSF shows that the c.5219+1G > T causes probably a frame shift, giving rise to a truncated protein with a loss of a large encoding fragment of FVIII protein. Although mRNA analysis for this novel mutation has not been carried out, this approach would be required, in the future, to elucidate the exact defect of this mutation in splicing.
Missense mutations: prediction of the mutate protein using the Swiss Pdb Viewer shows that the c.2189G > A (p.Cys730Tyr) mutation substitutes a conserved Cys residue located within a factor IX-binding region (Lys707-Asp714). 29 This substitution disrupts the disulfide bond formation (Cys630-Cys711) leading to a destabilization of the FVIII A2 domain (Figure 2A). The Cys630-Cys711 is 1 of the 7 disulfide bonds found to be indispensable for proper secretion and function of FVIII protein. 30 Furthermore, this mutation could create a new hydrogen bond with the neighboring serine 628. This interaction may leave the slightly exposed Cys 630 (fractional accessibility = 0.42) free on the protein surface (Figure 2A).
Although in silico analysis is generally sufficient to predict deleterious effect of novel mutations, the definitive tool is considered to be functional assay in vitro expression of the mutant protein in cell culture.
Analysis of deleterious effect of the missense mutation c.200A > C, p.Lys67Thr that results in substitution of Lys, a polar acidic residue, by Thr, a basic one, is discussed subsequently. As shown in Figure 2B, this substitution does not disrupt the interactions of Lys 67 (48) with the neighboring Lys 31 (12) and does not affect the protein structure. The substitution of this residue by Thr could confer a change in charge to the region and alter local interactions (Figure 2B). Although this substitution has a poor prediction, the real magnitude of its effect could be elucidating using Retro-Transcription PCR analysis of ectopic FVIII mRNA. The severe form detected in this patient could be due to the combine effect of mutation on protein and splicing.
For the c.6545G > A (p.Arg2182His) mutation, although both Arg and His are basic AAs, their side chains are structurally dissimilar. Therefore, this mutation is likely to have an effect on protein structure.
Polymorphism: the variant c.3780G > C, p.Asp1260Glu detected in our study was first considered as a polymorphism. 31 However, Repesse et al reported the presence of the c.3780G>C in 3 patients with severe form of HA and considered it as disease-causing mutation. 16 Today, it is clear that this variation is not a disease-causing mutation. Indeed, Ogata et al have demonstrated with functional approach that missense mutation in B domain has no effect on the protein and is not responsible for HA. 32
Gene Mutations and Severity
The present study confirms the well-known correlation between the types of mutation and the severity of HA. The types of mutations found were in agreement with the results reported in other studies. 6 –8 As expected, most patients with a severe form carry a molecular defect predicting a null allele (inversion intron 22, inversion intron 1, nonsense, and splice site mutations), whereas missense mutations were found in patients with moderate form of the disease. The 2 missense mutations (c.2189G > A and c.200A > C) were found in patients with severe form. The severe phenotype associated with these 2 mutations is in accordance with previously reported genotype/phenotype correlation. Indeed, the reported mutation c.2188T > G (p.Cys730Gly) that substitutes the same Cys was also associated with a severe phenotype. 8 Furthermore, the finding that the mutation c.200A > C is associated with a severe phenotype is in accordance with what was reported in the HAMSTeRS database. 6
Gene Mutations and Inhibitor Development
In this study, inhibitors were detected only in 12.5% (3 of 24) of patients with severe HA. This percentage is low when compared to previous reports, where development of inhibitors was predicted to occur in up to 30% of the patients with severe HA. 33 This relatively low incidence of inhibitors could be due to the fact that these patients did not receive frequent commercial replacement products, and they were treated only on request.
The type and location of the F8 gene mutation is an important determinant of inhibitor development in patients with severe HA. In a recent study, it was shown that mutations resulting in some protein synthesis were associated with a lower risk of inhibitor development, whereas mutations causing complete absence of protein were associated with a higher risk of inhibitors. 34 In our study, 2 severe patients with HA developed inhibitors at low levels (<5 BU), one had the nonsense mutation (c.322A > T) and the other had the variation (c.3780C > G).
Only 1 patient with intron 22 inversion developed inhibitor at high level (≥5 BU). This discordance in related patients (family 6) and in intron 22 inversion subgroup indicated additional factors other than F8 genotype for inhibitor formation. The pathogenesis of inhibitor formation still partly understood, several factors have been suggested to modulate this complication. The incidence of inhibitors depends on both genetic factors (type of mutation, ethnicity, family history of inhibitor, and HLA genotype) and nongenetic factors (age at first treatment, intensity of treatment, and multiple product switches). 35 –37
In conclusion, this is to our knowledge the first comprehensive report regarding HA mutations in Algeria where two novel mutations were identified. Despite the small number of patients reported in this article, they were the only ones who accepted and gave informed consent to participate in this study.
These initial promising observations would certainly pave the way for a large-scale study that involves a most important size of studied population to determine the distribution of these mutations in Algeria. Mutation analysis will contribute to facilitate genetic counseling and help in the prediction of the risk of inhibitor development.
Footnotes
Acknowledgments
The authors wish to thank patients and hemophilia care staff. We are indebted to Dr Le Bonniec Bernard for the precious help.
Authors’ Note
MA participated in samples collecting, molecular genetic studies, molecular modeling, and wrote the article. FZF was the primarily responsible for this work, designed the research, analyzed data, and corrected the article. MF, MSA, NSM, and AB gave intellectual support and discussion on the article. HT participated in the clinical diagnosis and sample collecting. CC participated in molecular genetic studies and gave intellectual support and discussion on the article. All authors read and approved the final article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The study was funded by the Laboratory of Molecular and Cellular Genetics, University of Science and Technology of Oran - Mohamed Boudiaf and the Algerian National Agency for the Development of Health Research (ANDRS).