
Abstract
The purpose of this study was to characterize differences in the prevalence of hereditary and acquired thrombophilia in patients with splanchnic vein thrombosis (SVT). A total of 88 consecutive patients with SVT, including Budd Chiari Syndrome (n = 47) and portal extrahepatic portal vein obstruction (n = 41), underwent comprehensive thrombophilia testing, including testing for heritable and acquired causes. In 33 (37.5%) patients, etiology could be explained by at least 1 of the heritable etiologic factors, and 31 (35.2%) patients could be explained by at least 1 of the acquired causes studied. The combination of multiple concurrent factors was present in 9 (11.4%) patients. Among the heritable causes, the risk of SVT was found increased in the presence of thrombophilia resulting from the deficiencies of the naturally occurring anticoagulant proteins, and the acquired thrombogenic factors were significantly associated with causation of thrombosis in adult patients with SVT.
Keywords
Introduction
The term splanchnic vein thrombosis (SVT) encompasses Budd-Chiari syndrome (BCS) and extrahepatic portal vein obstruction (EHPVO) besides portal vein thrombosis due to intrahepatic causes, mesenteric, and splenic vein thrombosis. 1 In both the disorders, pathogenesis is largely dependent on the presence of systemic prothrombotic conditions that promote thrombus formation in the respective hepatic vessels. 2 Current advances in noninvasive vascular imaging allow for the identification of chronic or asymptomatic forms.
The annual incidence of BCS and isolated mesenteric vein thrombosis is less than 1 per million individuals, while the incidence of EHPVO is about 4 per million; autopsy studies, however, suggest higher numbers. 1,3
The clinical presentations and risk factors for BCS and EHPVO are often shared. These risk factors may be inherited, like the protein C (PC), protein S (PS), and antithrombin III (ATIII) deficiency and factor V Leiden (FVL) mutation, or acquired—for example, paroxysmal nocturnal hemoglobinuria (PNH), Janus kinase 2 (JAK2) V617F mutations, and antiphospholipid antibodies. The detection of these defects could lead to relevant modifications in clinical treatment and patient management.
The data of etiologic factors in SVT are limited in Indian population. There are few studies from India that have evaluated the association of both hereditary and acquired thrombophilic factors in the causation of SVT. 4,5 Our objective was to find the role of thrombophilia testing in the causation of thrombosis in BCS and EHPVO, 2 of the most clinically relevant SVTs.
Materials and Methods
Patients/Study Population
Inclusion Criteria
This is a cross-sectional study of 88 consecutive patients with SVT who underwent comprehensive thrombophilia evaluation at the Department of Hematology, All India Institute of Medical Sciences, New Delhi, India, between August 2012 and March 2013. Budd-Chiari syndrome and EHPVO were diagnosed according to the previously published criteria. 6 Budd-Chiari syndrome was defined as hepatic venous outflow obstruction at any level from the small hepatic veins to the junction of the inferior vena cava and the right atrium, regardless of the cause of obstruction. Outflow obstruction caused by hepatic veno-occlusive disease and cardiac disorders is excluded from this definition. Extrahepatic portal vein obstruction was defined as obstruction of the extrahepatic portal vein. The obstruction may occlude the intrahepatic portal veins, splenic veins, or superior mesenteric veins. Extrahepatic portal vein obstruction also included the formation of portal cavernomas and the development of portal hypertension, which are associated with long-term disease. Isolated occlusion of the splenic or superior mesenteric vein, as well as portal vein obstruction associated with chronic liver disease or tumor, was not defined as EHPVO. 3,6
In all patients, the diagnosis was confirmed by imaging techniques including computed tomography, duplex ultrasonography, venography, or contrast-enhanced magnetic resonance imaging.
Exclusion Criteria
Patients with hepatocellular carcinoma, other overt or occult malignancies, liver cirrhosis, previously diagnosed myeloproliferative neoplasm (MPN), and those diagnosed with an overt MPN at the time of thrombosis were excluded. Chronic liver disease due to alcoholism, chronic viral hepatitis B or C, autoimmunity, iron overload, Wilson disease, and α-1 antitrypsin deficiency were also excluded.
The control group consisted of 90 healthy age and sex-matched controls patients who had undergone thrombophilia workup at coagulation laboratory during the same period.
Laboratory Testing
The distribution of number of patients and the tests performed are given in Table 1. For thrombophilia testing, 9 volumes of blood were collected in 1 volume of 3.18% trisodium citrate from patients and controls. The citrated blood sample was spun at 3500 rpm for 10 minutes at 4°C. The plasma was aliquoted immediately and preserved at −70°C until use. All the plasma samples were tested within 2 months of blood collection.
Age and Gender Distribution of Patients With SVT.
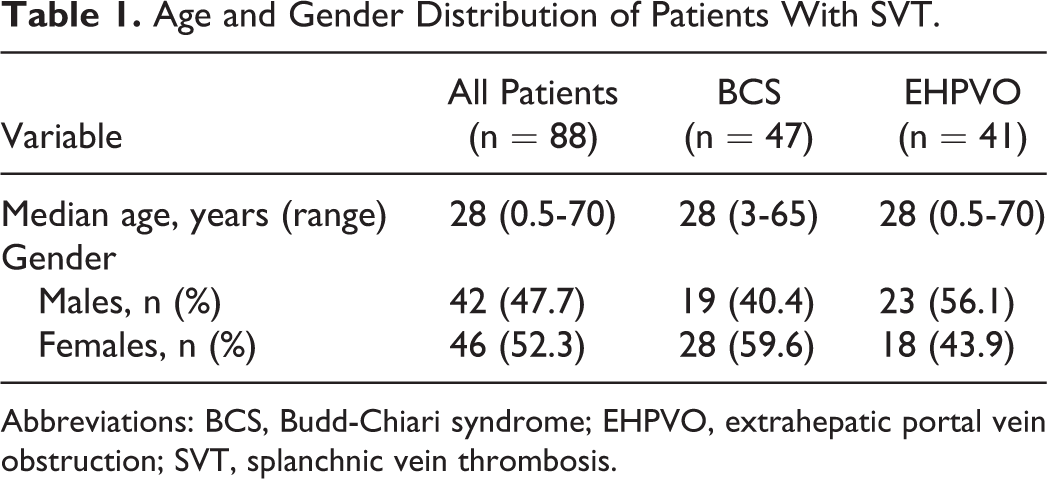
Abbreviations: BCS, Budd-Chiari syndrome; EHPVO, extrahepatic portal vein obstruction; SVT, splanchnic vein thrombosis.
Protein C and PS antigen activity was determined by sandwich enzyme-linked immunosorbent assay using commercially available kits (Corgenix, Inc, Colorado, USA). The expected normal activity of PC per kit definition was 72% to 130% and that of PS was 60% to 135%. The quantification of ATIII was done using STACHROM ATIII kit on an automated STA Compact Analyzer (Diagnostica Stago, France). Normal value for ATIII ranged from 80% to 120%. Activated protein C resistance (APCR) was performed by 2 methods. In 40 patients, the APCR ratio was determined by the clotting time obtained in the presence of added human APC divided by the activated partial thromboplastin time in the absence of APC. For this study, the cutoff ratio used to determine positive APCR was a value of ≤2.1 seconds. In 25 patients, APCR was performed using the Stago Diagnostica kit (Diagnostica Stago). Results were considered positive for cutoff values of less than 120 seconds. Detection of lupus anticoagulant was performed by 2 methods, first by the kaolin clotting time method 7 and second using the STACLOT LA kit (Diagnostica Stago). Anti-β2 glycoprotein I (anti-β2GPI) immunoglobulin (Ig) G/IgM antibodies were quantitatively determined by immunometric enzyme immunoassay (EIA; ORGENTEC Diagnostica GmBH; normal range 2-8 U/mL). Serum homocysteine levels were measured by modified EIA method (Diazyme Laboratories, San Diego, California; normal range 5-15 μmol/L). All samples showing borderline results were repeated, and only those samples that were consistent were considered as deficient.
Total genomic DNA was isolated from peripheral blood leukocytes using commercially available kit (BioServe, USA). Mutational analysis for FVL was performed in patients who tested positive for APCR or in those where borderline results for APCR were obtained. Also, patients who were on oral anticoagulants and could not be worked up for APCR were subjected to FVL mutation analysis. Mutational analysis for FVL and prothrombin G202010A was performed by polymerase chain reaction (PCR) restriction fragment length polymorphism method. 8 The existence of C677T methylene tetrahydrofolate reductase (MTHFR) polymorphism was detected by PCR, restriction enzyme digestion, and gel electrophoresis. JAK2 V617F mutations were analyzed and determined by Tetra Primer Amplification Refractory Mutation System PCR technique, as described previously. 9 Blood of 2 mL was collected in EDTAtubes for complete blood count, and PNH clone was quantitated on flow cytometric analysis using monoclonal antibodies against CD55 and CD59 on peripheral blood leucocytes.
Statistical Analysis
Comparison between categorical variables was performed with chi-square testing and between continuous variables with the Student t test. Odds ratios (ORs) for SVT associated with various thrombophilia markers and corresponding 95% confidence intervals (CIs) were calculated using logistic regression, adjusted for age and sex. P values were 2 tailed, and statistical significance was set at P < .05.
Results
Demographic Characteristics
A total of 88 patients with SVT including 47 (53.4%) patients witth BCS and 41 (46.6%) patients with EHPVO underwent comprehensive thrombophilia testing. The median age of the patients was 28 years (range 6 months to 70 years). Females outnumbered males (46 females, 52.3%; Table 1). Patients were young with 23 (26.1%) patients being less than 18 years of age and 78 (88.6%) patients less than 45 years of age. Three female patients had additional known circumstantial risk factors as they presented with BCS in postpartum period. None of the patients had associated family history of thromboses.
Thrombophilia Workup
Protein C was the commonest coagulation inhibitor found deficient in patients with both BCS and EHPVO (n = 19 of 69; 27.5%; OR: 33.8; 95% CI, 4.3-250.8, P = .0007) across both the age groups (Tables 2 and 3). Antithrombin III was the second commonest coagulation inhibitor found deficient (n = 7 of 69; 10.1%). Antithrombin III deficiency was found only in patients with BCS (chi-square test, P = .006), while the PS deficiency was commoner in patients with EHPVO; however, this difference was not statistically significant (chi-square test, P = .154). Concurrent deficiencies of PC, PS, and ATIII were found in 1 patient, while 2 other patients were deficient in both PC and PS.
Results of Thrombophilia Testing in Patients With BCS and EHPVO.
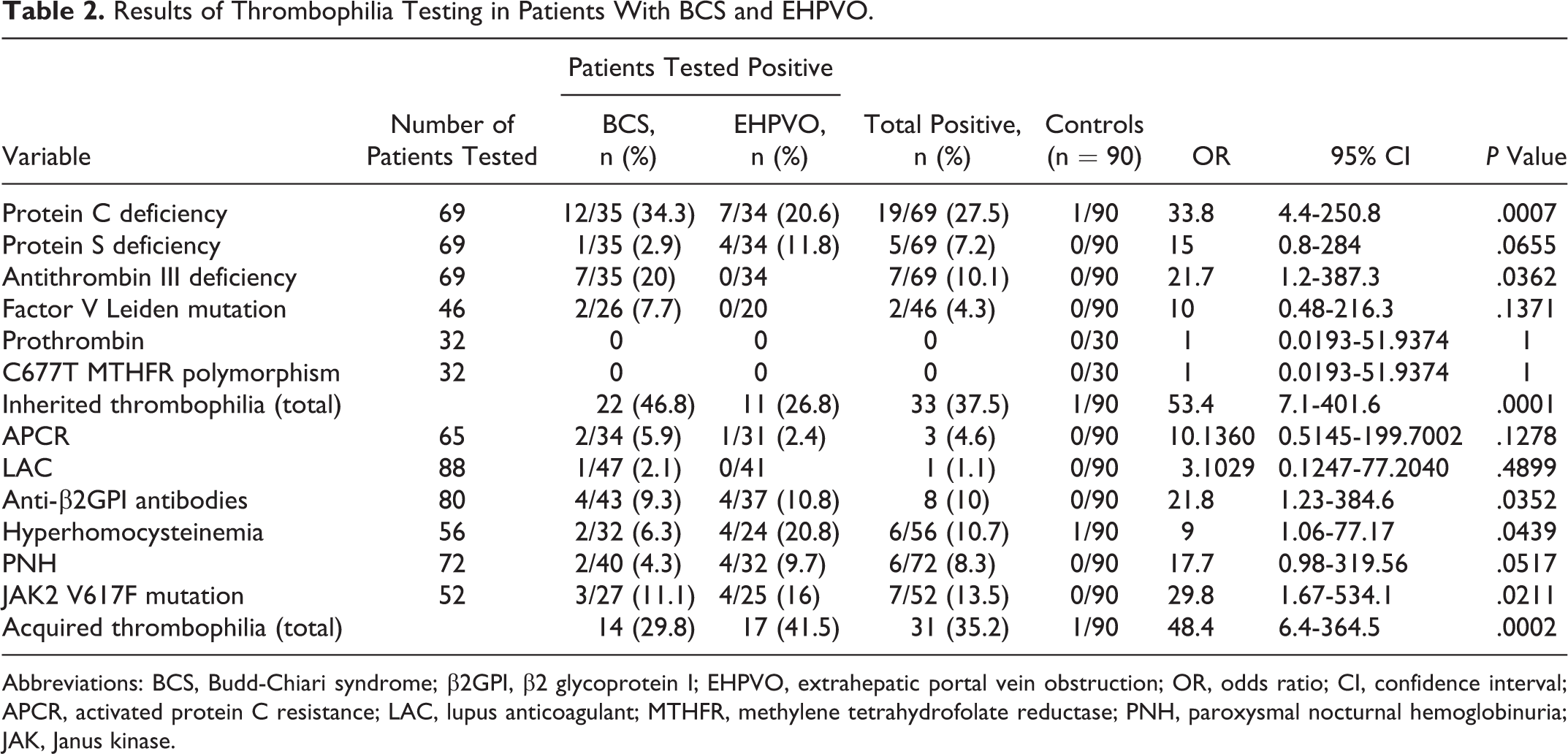
Abbreviations: BCS, Budd-Chiari syndrome; β2GPI, β2 glycoprotein I; EHPVO, extrahepatic portal vein obstruction; OR, odds ratio; CI, confidence interval; APCR, activated protein C resistance; LAC, lupus anticoagulant; MTHFR, methylene tetrahydrofolate reductase; PNH, paroxysmal nocturnal hemoglobinuria; JAK, Janus kinase.
Distribution of Patients With Respect to Age Group and Causative Factors.
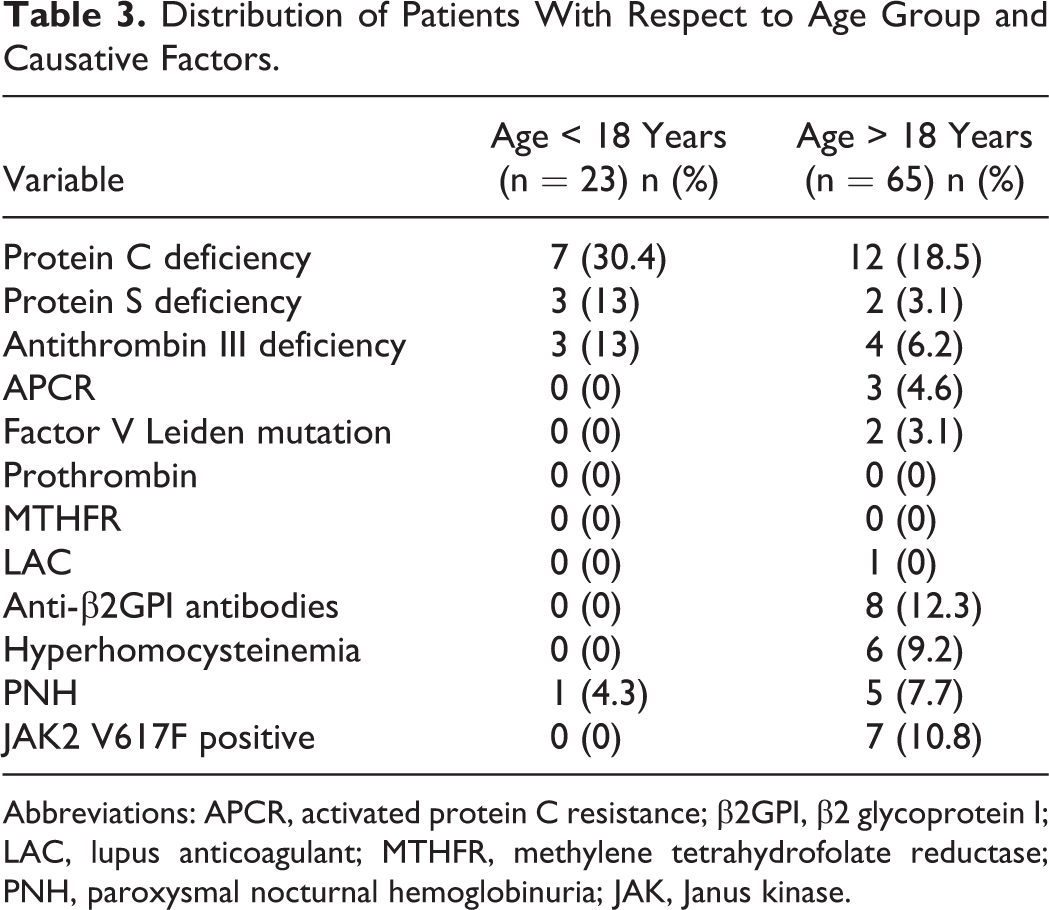
Abbreviations: APCR, activated protein C resistance; β2GPI, β2 glycoprotein I; LAC, lupus anticoagulant; MTHFR, methylene tetrahydrofolate reductase; PNH, paroxysmal nocturnal hemoglobinuria; JAK, Janus kinase.
The APCR was found in 3 (4.6%) patients, and of these, heterozygous FVL mutation was seen in 2 (2.3%) patients. In the third patient with APCR positivity, results of FVL mutation testing were not available. None of the patients worked up in our study showed C667T MTHFR polymorphism or prothrombin gene mutation.
JAK 2V617F mutation was the commonest acquired cause for thrombophilia found in 7 (13.5%) of the 52 patients in the absence of an overt MPN. Hyperhomocysteinemia was seen in 6 (10.7%) of the 56 patients. None of the patients with hyperhomocysteinemia had associated MTHFR polymorphism. Anti-β2GPI antibodies were found in 8 (10%) patients. Paroxysmal nocturnal hemoglobinuria clone was positive in 6 (9.7%) patients, and the clone size was more than 50% in 1 of these patients. Two of the PNH-positive patients had associated hyperhomocysteinemia.
A gender-wise distribution of patients was made for the present study, and anti-β2GPI antibodies were more often found in females, while PNH positivity was more common in males (Table 4); these differences were not statistically significant (P > .05).
Distribution of Patients With Thrombophilic Disorders on the Basis of Gender.

Abbreviations: APCR, activated protein C resistance; LAC, lupus anticoagulant; MTHFR, methylene tetrahydrofolate reductase; PNH, paroxysmal nocturnal hemoglobinuria; JAK, Janus kinase.
The acquired causes of thrombophilia, that is, anti-β2GPI antibodies, acquired APCR, hyperhomocysteinemia, PNH positivity, and JAK2 V617F mutation positivity in SVT, were significantly higher in adult patients (older 18 years of age) as compared to children and adolescents (P = .001; Table 4).
The combination of multiple concurrent factors was present in 9 (10.2%) patients with SVT, 5 each of BCS and EHPVO, respectively (Table 5). Overall 33 (37.5%) patients could be explained by at least one of the heritable etiologic factors, and 31 (35.2%) patients could be explained by at least 1 of the acquired causes studied.
Multiple Concurrent Thrombophilic Factors in SVT.
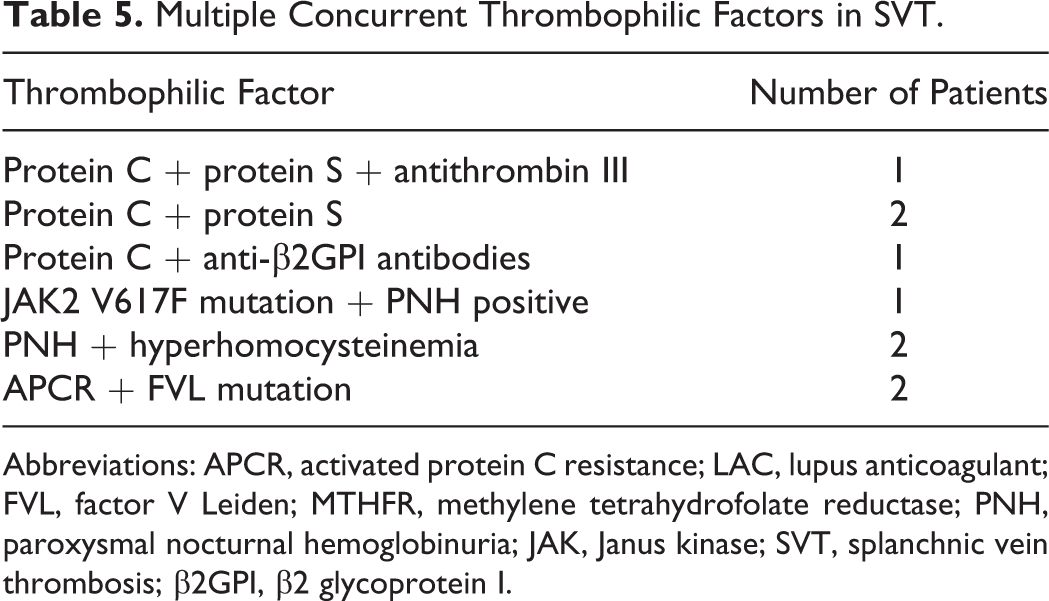
Abbreviations: APCR, activated protein C resistance; LAC, lupus anticoagulant; FVL, factor V Leiden; MTHFR, methylene tetrahydrofolate reductase; PNH, paroxysmal nocturnal hemoglobinuria; JAK, Janus kinase; SVT, splanchnic vein thrombosis; β2GPI, β2 glycoprotein I.
Hereditary causes were more often associated with thrombosis in patients with BCS as compared to patients with EHPVO (46.8% and 29.2% respectively, P = .09).
Discussion
Risk factors for SVT vein thrombosis can be local or systemic, and the latter is influenced by inherited or acquired conditions. This is the first study that has evaluated a spectrum of thrombogenic factors implicated in the pathogenesis of SVT. One or several of these prothrombotic disorders were found in 36 (76.6%) of the 47 patients with BCS and in 28 (68.3%) of the 41 patients with EHPVO.
Diagnosis of inherited deficiencies of PC, PS, and AT in patients with BCS and EHPVO needs to be carefully interpreted as acquired deficiencies of coagulation factors and their inhibitors take place due to liver dysfunction in these disorders. These acquired deficiencies can occur due to reduced synthetic capacity, oral anticoagulation, or due to the utilization of these factors during acute episodes. Levels of PS remain substantially high even in patients with liver dysfunction owing to extrahepatic synthesis of PS. 10,11 Janssen et al showed that among the 3 factors, PC is the only significantly associated factor with both PVT and BCS, respectively. 6 To minimize the number of incorrectly diagnosed inherited deficiencies, we excluded patients with features of chronic liver disease and deferred testing for 3 months for those on anticoagulant therapy and/or during acute thromboses. In all of our patients with PC and PS deficiencies, an acquired deficiency was ruled out by normal liver function test results and a normal prothrombin time. Inherited deficiencies were found to be commoner in younger patients in our study.
The prevalence of FVL mutation in patients with BCS and PVT has been found to range between 6.8% to 31.8% and 2.3% to 16.7% in patients with BCS and PVT, respectively, in various studies reported in the literature. 10 In Asia, particularly in India, the prevalence of FVL mutation is very low both in the general population and in patients with DVT. 4,5,12,13 In the present study, the prevalence of FVL mutation was seen only in 2 cases, and both were heterozygotes suggesting additional acquired factors in pathogenesis. Both the patients with FVL mutation showed APCR. The third patient with APCR did not undergo FVL mutation screening and had borderline values of PS, that is, 70%. Hence, it is probable that the APCR may be acquired and not hereditary, as previous studies have suggested that PS and PC levels may affect acquired APCR. 14 Factor V Leiden was not implicated as a major factor in SVT in the present study, unlike other studies in the literature. 15 This can be attributed to the small sample size of patients who underwent testing for this mutation. Similarly, the complete lack of MTHFR C677T polymorphism could also be attributed to the small sample size.
In India and many other Asian countries, prothrombin mutation has not been detected, and screening for this mutation is generally not indicated in these populations. 16 –18 None of our tested patients or controls showed this mutation, and further screening was thus abandoned.
The association of BCS and EHPVO with antiphospholipid antibodies has been documented in several case studies 19 –21 and reports. 22,23 In our study, anti-β2GPI antibodies were the only demonstrable etiologic agent in 7 patients, and 1 patient had associated JAK2 V617F mutation. This suggests a strong association of anti-β2GPI antibodies with SVT. The current consensus is that a second hit is necessary to unveil the prothrombotic activity of antiphospholipid antibodies. 24 Paroxysmal nocturnal hemoglobinuria is a well-recognized cause of BCS and PVT 25,26 and was found positive in 6 patients in our study.
Hyperhomocysteinemia is a relatively weak risk factor for both arterial and venous thrombosis. 10,27 Hyperhomocystenemia was found in 6 patients in our study, and all of them were >18 years of age. This may suggest the role of other comorbid conditions causing rise in homocysteine levels, family history of cardiovascular disease, hypertension, diabetes, and hyperlipidemia.
Several studies have shown that JAK2V617F detection is warranted for diagnosing MPD in patients with BCS or PVT, and the prevalence of this mutation has been reported to be as high as 75% among patients with prior or overt diagnosis of MPN and 26% among those without overt MPN. 28 –30 The present study brought out 7 (13.5%) of the 52 positive results from our center. None of these patients had overt MPN. This result further suggests that the search for JAK2 V617F mutations associated with CMPD is warranted in patients with BCS and EHPVO, because they can establish the clonal nature of the disease even in the absence of an overt MPN.
The only limitation of the present study was that all patients could not be evaluated for every thrombogenic defect. This was mainly due to economic constraints as many of our patients could not afford expensive molecular mutation testing. As already mentioned in methodology, FVL testing was carried out in patients positive for APCR or those on oral anticoagulants, where APCR could not be tested. Nevertheless, important conclusions have been drawn from this study.
Conclusion
Thus, overall 33 (37.5%) patients could be explained by at least 1 of the heritable etiologic factors and 18 (20.4%) patients could be explained by at least 1 of the acquired causes studied. The combination of multiple concurrent factors was present in 9 (11.4%) patients. Among the heritable causes, the risk of SVT was found increased in the presence of thrombophilia resulting from the deficiencies of the naturally occurring anticoagulant proteins. The acquired thrombogenic factors, that is, JAK2 V617F mutations, PNH, hyperhomocysteinemia, and anti-β2GPI antibodies were causative more often in adult patients with SVT. Thus, a complete hematological workup, including diagnosis of inherited and acquired prothrombotic factors, should be performed in BCS and EHPVO, irrespective of whether 1 prothrombotic factor has already been identified. This is in particular relevant for identifying MPN, which is also often accompanied by other prothrombotic factors, so that additional treatment modalities if required can be instituted.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.