
Abstract
Background: The purpose of this study is to investigate the acute and chronic effects of cigarette smoking on cyclooxygenase- 1(COX-1)-mediated platelet reactivity among cigarette smokers. Methods: The levels of collagen-induced platelet aggregation, platelet COX-1 activity, and expressions were compared between smokers and age-matched nonsmokers. In smokers, the acute effects of cigarette smoking were assessed by repeating these measurements an hour after smoking. Results: Twenty-five smokers and age-matched nonsmokers (all men; mean age, 29 years) were studied. Collagen-induced platelet aggregation and plasma/urinary thromboxane B2 (TXB2) and 11-dehydroxythromboxane B2 levels were higher in cigarette smokers compared to nonsmokers. Greater expression of platelet COX-1 was observed in smokers than in nonsmokers. Among smokers, collagen-induced platelet aggregation correlated positively with platelet volume and circulating nicotine and cotinine concentrations. The levels of plasma/urinary TXB2 were significantly increased an hour after cigarette smoking. Conclusion: Cigarette smoking aggravates COX-1-mediated platelet reactivity in young, otherwise healthy, smoking men.
Introduction
The deleterious effects of cigarette smoking is less recognized in young adults. Epidemiology data indicate unexpectedly high cardiovascular events in young individuals who, because of their age, are not typically considered to have increased cardiovascular risk. 1 Among adults who had myocardial infarction (MI), a higher prevalence of cigarette smokers was observed in patients ≤40 years compared to those >60 years. 2 Smokers below the age of 50 are 5 times more likely to die from MI than nonsmokers. 3 Despite being at increased risk of cardiovascular events, limited data are available on the changes in platelet reactivity among young cigarette smokers.
Platelets are central to the pathogenesis of atherosclerosis and arterial thrombosis. 4 During early atherogenesis, platelets adhere to dysfunctional endothelium through minor fissuring of the atheromatous plaque and contribute to the growth and propagation of these plaques. Occlusion of the artery, either by progressive narrowing or by acute occlusion instigated by plaque rupture, may trigger the onset of cardiovascular events. The reactivity of platelets and the ability of the platelets to aggregate are regulated by various mediators, especially those involved in the cyclooxygenase-1 (COX-1) pathway. The COX-1 mediates the synthesis of thromboxane A2 (TXA2), an arachidonic acid (AA) metabolite formed by the combined catalytic actions of and thromboxane A synthase (TXAS). 5 These act via the platelet TXA2 receptor that initiates changes in platelet morphology and subsequently induces platelet aggregation. 5 Several studies have observed alterations in platelet aggregation and changes in COX products in unselected cohorts of cigarette smokers. 6 –10 These studies, however, were limited by the choice of markers to measure cyclooxygenase activities, the lack of suitable controls, and the limited information on the temporal changes of these markers in relation to cigarette smoking. Although COX-1 is found to be more abundantly expressed than COX-2 in platelets, 11 the changes in the activities of COX-1 pathway are poorly understood in young smokers.
The purpose of this study is to assess the acute and chronic effects of cigarette smoking on markers of COX-1-mediated platelet reactivity in a cohort of young, otherwise healthy, men. We chose to recruit men since cigarette smoking was significantly more prevalent in men compared to women in Singapore (24.7% vs 4.2%). 12 We hypothesize that platelet COX-1 activities are increased in young smokers and that acute and chronic smoking exerts distinctive effects on COX-1-mediated platelet activities in these individuals. Platelet aggregation (induced by collagen), plasma and urinary thromboxane B2 (TXB2), and platelet COX-1 expression/activity were studied in smokers and nonsmokers. In smokers, these markers were compared 8 hours following cigarette abstinence and an hour after smoking.
Materials and Methods
Participants and Study Design
Adult men above the age of 21 who were not previously diagnosed with hypertension, diabetes mellitus, or hyperlipidemia were studied. Participants who consumed medications (including antiplatelet therapy) within a month prior to their study involvement were excluded. Smokers were defined as those who smoked cigarettes daily for at least 5 years. Nonsmokers comprised age-matched men who had never smoked or exposed to cigarette smoke in their home/work environments. Clinical information (including demographic, body mass index [BMI], blood pressure, and cigarette consumption) was obtained using structured questionnaires. Power calculations, performed a priori on the primary variables (plasma thromboxane B2) derived from data of a pilot study, indicated that a minimum sample size of 25 participants was required in each group with a probability (power) of .80. The type I error P value was .05 (α).
The chronic effects of cigarette smoking were compared between smokers and age-matched controls (nonsmokers). Blood and urine samples for analysis were collected from the participants 8 hours following fasting and cigarette abstinence. Among smokers, the immediate effects of smoking on platelet activity were examined by comparing the markers before and after cigarette smoking. A second set of blood and urine samples were collected an hour after cigarette smoking. Smokers were asked to smoke 1 cigarette of their own—the brand of cigarette was not standardized to reflect “real-world” situation.
Glucose, cholesterol, triglyceride, low-density lipoprotein (LDL), and high-density lipoprotein (HDL) concentrations were measured in serum using the Cobas c111 Photometric Analyzer (Roche Diagnostic GmbH, Mannheim, Germany). Platelet count, mean platelet volume (MPV), and platelet distribution width (PDW) were determined in each blood sample using the Sysmex pocH-100i Automated Hematology Analyzer (Sysmex Corp, Kobe, Japan). The study protocol was reviewed and approved by the Domain Specific Review Board, National Healthcare Group, Singapore, and each participant provided a written informed consent prior to their study participation.
Whole Blood Platelet Aggregation
Whole blood platelet aggregation was measured using the Multiplate Platelet Function Analyzer (Dynabyte GmbH, Munich, Germany), as described previously. 13
Peripheral Platelet Isolation and Stimulation of Platelets
Peripheral platelets were initially separated from whole blood by centrifugation at 200x g for 10 minutes. Platelet-rich plasma obtained was further purified by centrifugation at 600x g for 15 minutes. Platelet numbers, MPV, and PDW did not differ after isolation. The isolated platelets (1 mL of 100 × 106 cells/ mL in Hank's buffered salt solution with bovine serum albumin; HBHS) were incubated at 37°C for 5 minutes prior to COX stimulation. Platelets were then stimulated with calcium ionophore A23187 (2.5 µg/mL; Sigma Aldrich, Missouri) at 37°C for 15 minutes. Supernatant from the cell suspension was collected and stored at −80°C before TXB2 extraction and analyses. A portion of the platelets in HBHS was stored at −80°C for COX-1 expression measurements.
Thromboxane Metabolites, Nicotine, and Cotinine Measurements
Circulating COX metabolites, TXB2 and 11-dehydro thromboxane B2 (11-dhTXB2), were determined in plasma and urine using isotope-labeled gas chromatography–mass spectrometry (GC-MS). 14 Circulating plasma TXB2 and 11-dhTXB2 concentrations were presented uncorrected, while urinary TXB2 and 11-dhTXB2 concentrations were expressed per unit creatinine. Nicotine and cotinine levels were measured by GC-MS using previously described methods. 15,16
Platelet Cyclooxygenase-1
Cyclooxygenase-1 protein was isolated by suspending platelets in Radio-Immunoprecipitation Assay (RIPA) buffer containing protease inhibitor (Sigma-Aldrich P8340; 20 µL/mL of RIPA buffer) for 30 minutes in ice. The supernatant containing the proteins was obtained by centrifugation at 10 000 rpm for 10 minutes. The COX-1 expression was determined using electrophoresis on sodium dodecyl sulfate (SDS) gel and subsequent Western blot analysis. The COX-1 (70 kDa) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, 39 kDa) proteins were visualized with an ECL Western blot analysis system (Amersham, UK) and exposed to radiofilm (Amersham). The intensity of protein band on exposed film was measured using ImageJ software (The National Institutes of Health, USA). The COX-1 protein expression was expressed as the ratio of the intensity of protein band relative to GAPDH in the same membrane.
Statistical Analysis
Statistical analyses were performed using SPSS version 15.0 (SPSS Inc, Chicago, Illinois, USA). Mean ± standard deviation (SD) was used to describe normally distributed data, while median (interquartile range) was used to describe nonparametric data. For normally distributed data, between-group (nonsmokers vs smokers) and within-group (smokers before and after smoking) differences were analyzed using unpaired and paired t tests, respectively. Whereas for nonparametric data, between-group and within-group differences were analyzed using Mann-Whitney U unpaired and Wilcoxon paired tests, respectively. The chronic effects of cigarette smoking were compared between nonsmokers and smokers 8 hours following cigarette abstinence. The acute effects of cigarette smoking were examined in smokers an hour after cigarette smoking. Spearman correlation analyses were performed to investigate the associations between the study covariates. Statistical significance was set at P < .05.
Results
Participant Characteristics
A total of 25 men smokers of mean age 29.4 ± 4.5 and 25 men nonsmokers of mean age 28.5 ± 5.1 were included. All participants are of Asian ethnicity. On average, smokers had smoked an average of 7.2 ±± 3.7 cigarettes a day for 13.0 ± 7.2 years (cigarette pack years, 4.2 ± 2.6). There were no significant differences in age, BMI, blood pressures, as well as serum levels of glucose, LDL, HDL, triglyceride (TG), and total cholesterol (TC) between smokers and nonsmokers (Table 1). None of the study participants were diagnosed with hypertension, hyperlipidemia, or diabetes mellitus. 17 Nicotine and cotinine concentrations in blood plasma and urine, expectedly, were significantly higher in smokers compared to nonsmokers (Table 1). Cigarette smoking acutely increased nicotine and cotinine levels in plasma and urine of smokers (Table 1).
Participant Characteristics.a
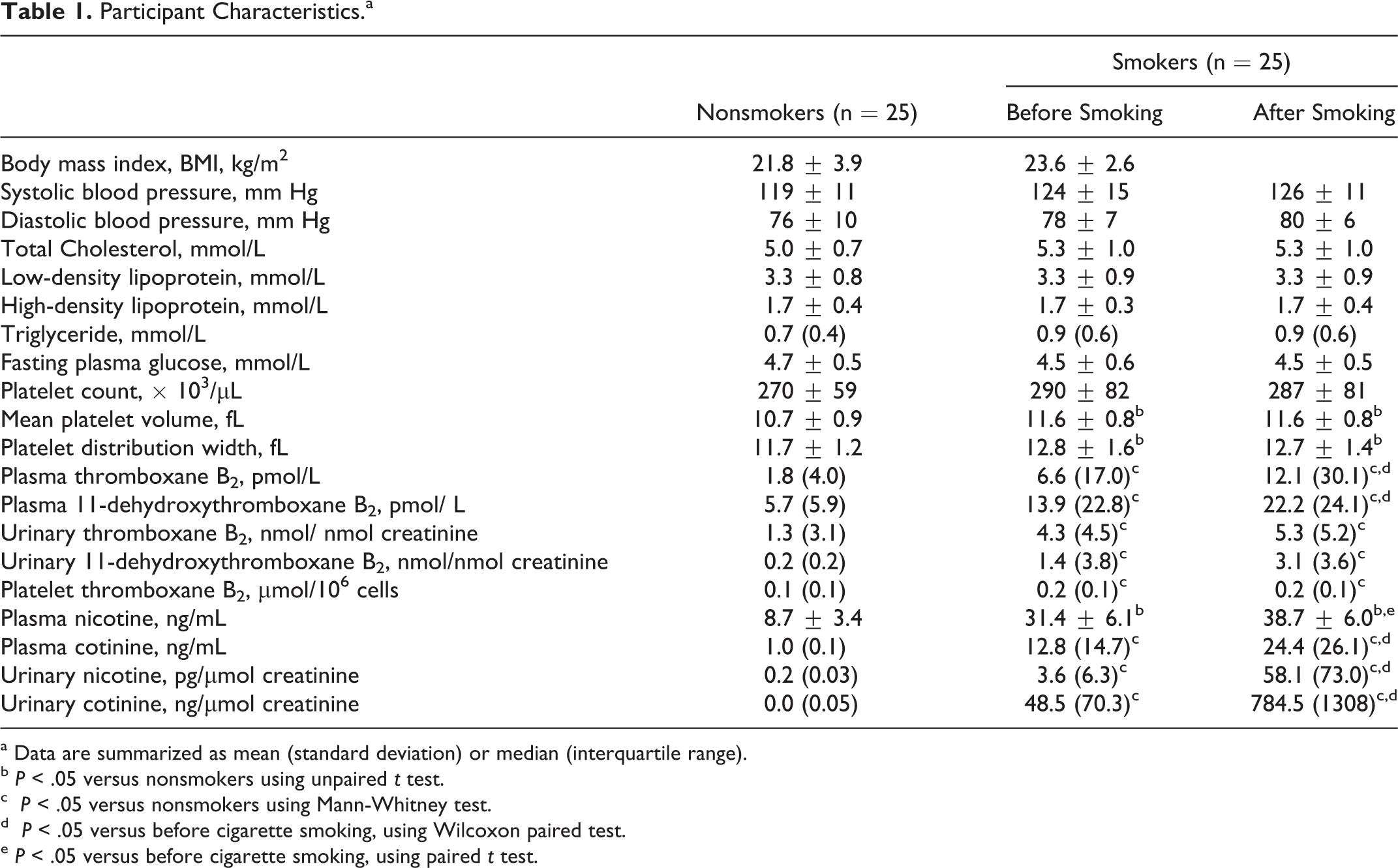
a Data are summarized as mean (standard deviation) or median (interquartile range).
b P < .05 versus nonsmokers using unpaired t test.
c P < .05 versus nonsmokers using Mann-Whitney test.
d P < .05 versus before cigarette smoking, using Wilcoxon paired test.
e P < .05 versus before cigarette smoking, using paired t test.
Platelet Count, Mean Platelet Volume, and Ex Vivo Platelet Aggregation
Despite having comparable platelet counts, smokers had significantly higher levels of PDW and MPV (indicative of larger platelet size) compared to nonsmokers (Table 1); these levels were unchanged an hour following cigarette smoking (Table 1). The extent of platelet aggregations was measured following ex vivo stimulation of collagen. After 8 hours of cigarette abstinence, smokers had significantly higher collagen-induced platelet aggregation than nonsmokers (Figure 1). The levels of collagen-induced platelet aggregation tended to be higher following an hour after cigarette smoking, but these did not differ from the baseline levels (Figure 1).
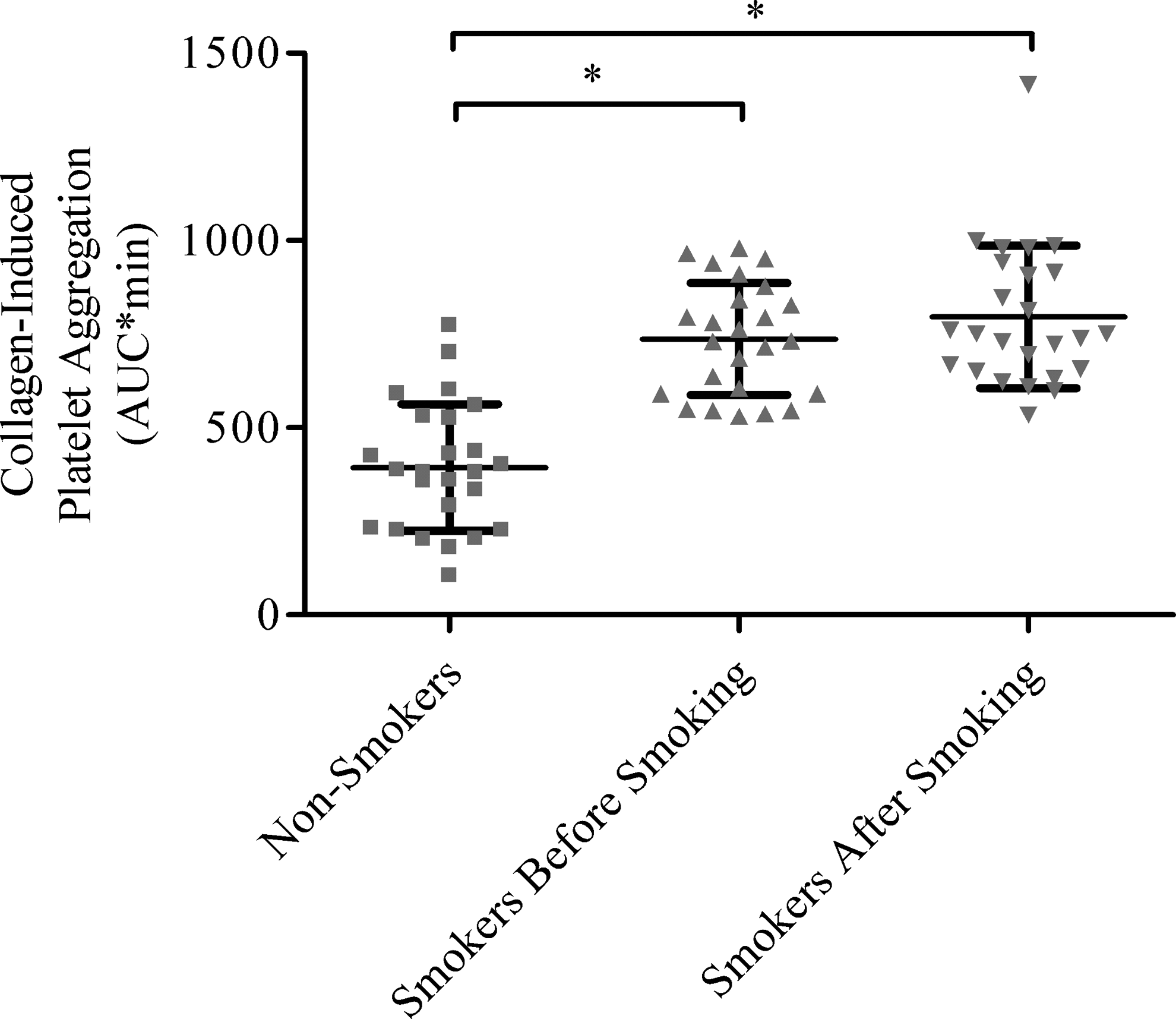
Platelet aggregation in whole blood from nonsmokers and smokers. Platelet aggregation (mean ± standard deviation, area under the curve*min) induced by collagen (2.5 µg/ mL) in whole blood of nonsmokers (N = 25) and smokers (N = 25) before and after cigarette smoking. *indicates P < .05 using unpaired t test.
Cyclooxygenase-1 Activities and Expression
The activities of COX enzymes were determined by measuring TXB2 and 11-dhTXB2 concentrations in plasma and urine. Compared to nonsmokers, smokers had significantly higher plasma and urinary levels of TXB2 and 11-dhTXB2 concentrations (all Ps < .05; Table 1).
Platelet COX activity was determined by measuring the production of TXB2 from stimulated platelets. Stimulated platelets isolated from smokers produced significantly higher amounts of TXB2 than those isolated from nonsmokers (P < .05; Table 1). Platelets isolated from smokers showed significantly higher COX-1 expression compared to nonsmokers (P < .05; Figure 2). Cigarette smoking acutely increased plasma and urinary TXB2 and 11-dhTXB2 concentrations (P < .05 for plasma TXB2 and 11-dhTXB2 only; Table 1) but did not alter the production of TXB2 in isolated platelets (Table 1) and platelet COX-1 expressions (Figure 2).
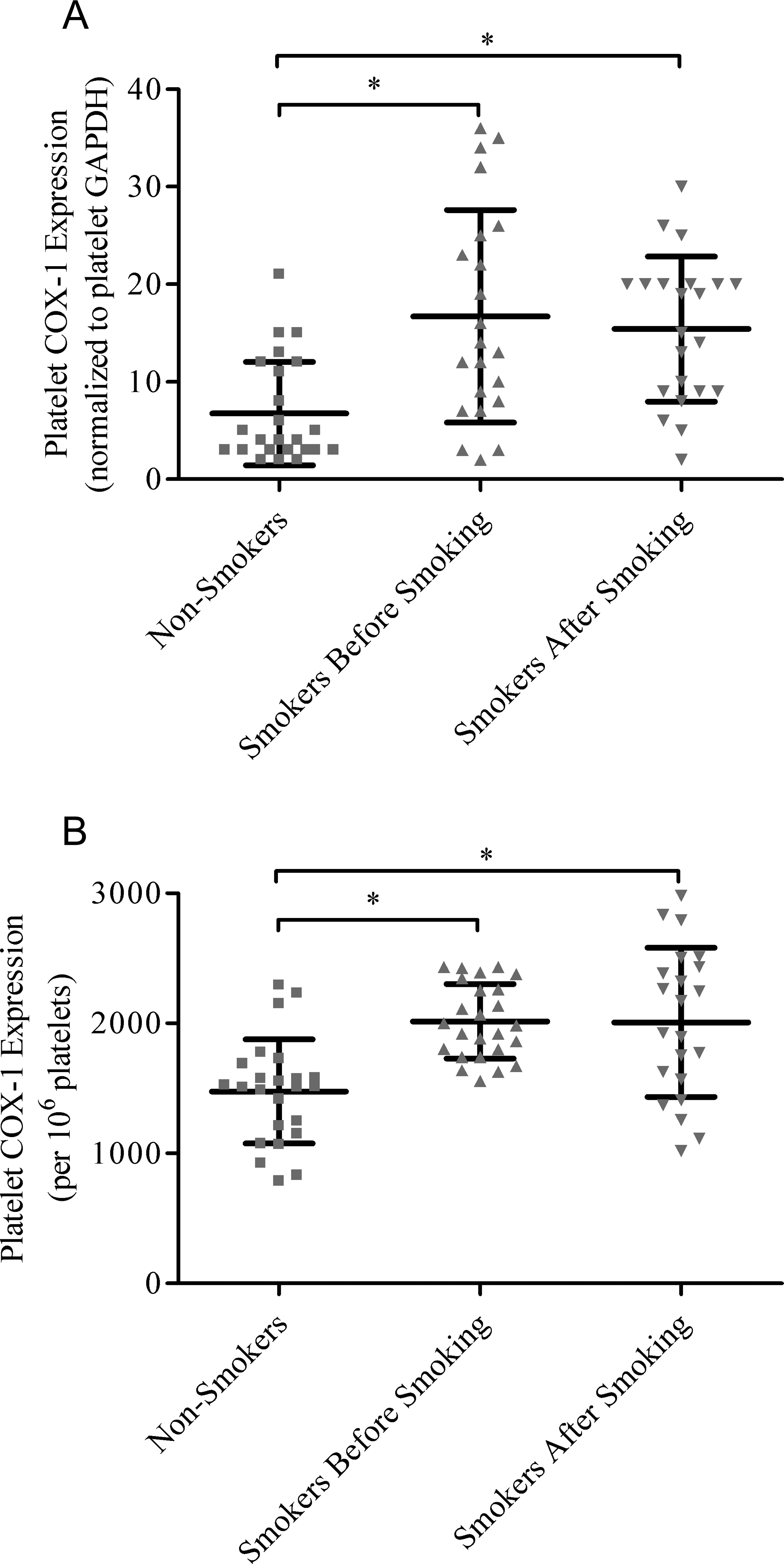
Platelet cyclooxygenase-1 expression. Expression of cyclooxygenase 1 (mean ± standard deviation) normalized to (A) glyceraldehyde 3-phosphate dehydrogenase and (B) 106 cells in isolated platelets of nonsmokers (N = 25) and smokers (N = 25) before and an hour after cigarette smoking. *indicates P < .05 using unpaired t test.
Correlation Analysis
Collagen-induced platelet aggregation correlated significantly with MPV, plasma TXB2, platelet TXB2 production, and platelet COX-1 expression (Table 2). The TXB2 concentrations in plasma correlated significantly with their urinary levels (nonsmokers, r = .85 and smokers, r = .89). Plasma TXB2 concentration and platelet TXB2 production correlated significantly with the COX-1 expression in these platelets (Table 2). Platelet aggregation (in response to collagen stimulation) correlated significantly with plasma nicotine and cotinine concentrations in smokers (Table 2). These significant correlations, however, were not observed an hour after cigarette smoking. Markers of COX-1 activities (measured in plasma, urine, and activated platelets) were not associated with BMI, LDL, HDL, TG, TC, and fasting glucose measurements, both in smokers and in nonsmokers.
Correlation Coefficients between Collagen-Induced Platelet Aggregation, Platelet Cyclooxygenase-1 Expression, and Other Measured Markers of Platelet Cyclooxygenase-1 Activity in Male Nonsmokers (N = 25) and Smokers Before Smoking (N = 25).
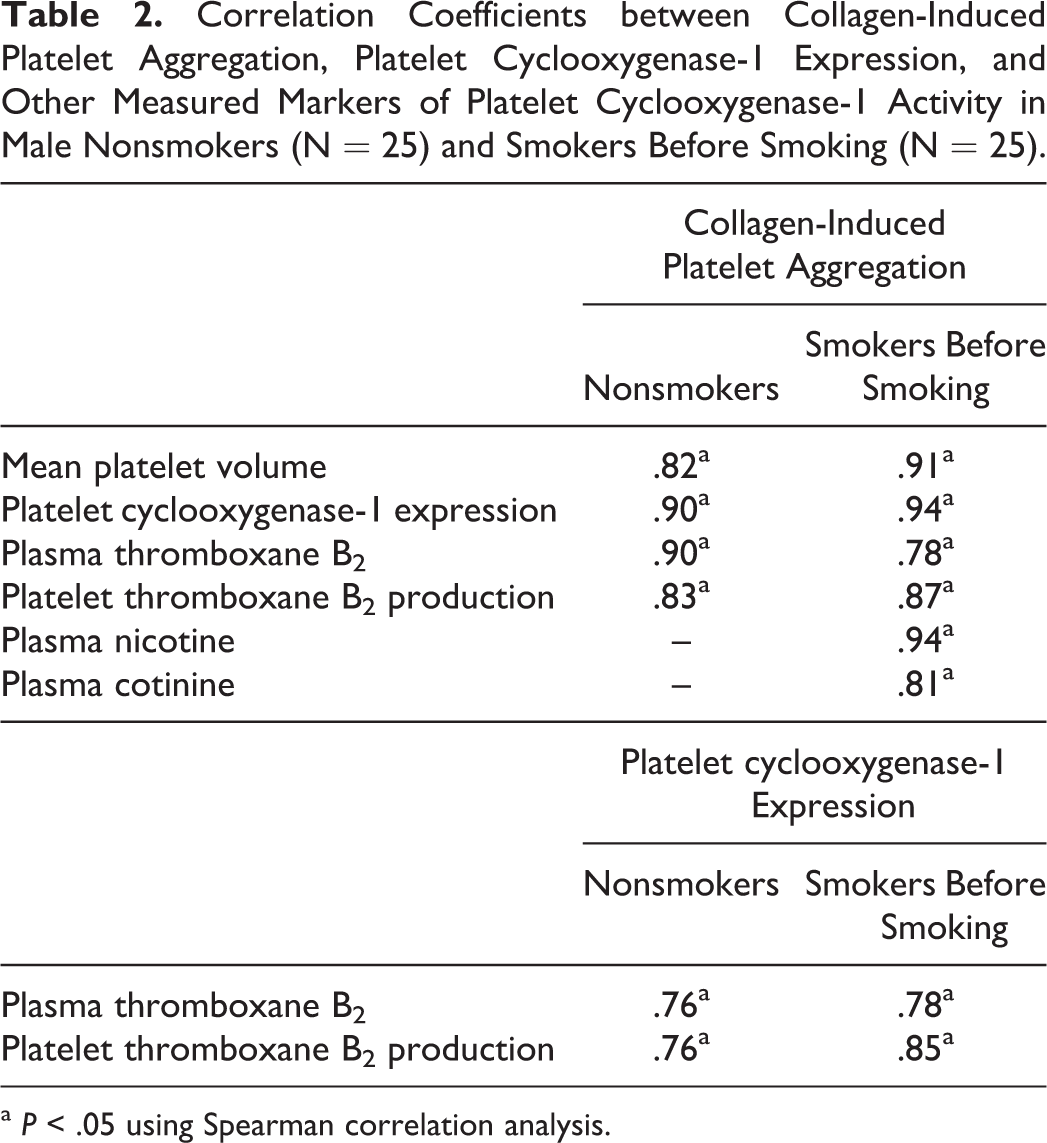
a P < .05 using Spearman correlation analysis.
Discussion
Cigarette smoking alters platelet function by modulating changes in platelet morphology, enzyme activities, and protein expressions. 7 Using whole blood electrical impedance platelet aggregation measurements, we observed increases in collagen-induced platelet aggregations among young, healthy smokers when compared to nonsmokers. Collagen-induced aggregation is used as a specific marker for COX activities. Collagen, produced during vascular injury, activates phospholipase A2, allowing the hydrolysis of AA from membrane phospholipids, the first step in the pathway toward the release of TXA2. The rise in platelet aggregation was accompanied by an increase in platelet volume, metabolites of COX activities, and expression of COX-1 in activated platelets. Previous studies that investigated the effects of cigarette smoking in platelet aggregation had produced conflicting findings. 6,18 In a study that examined the effects of smoking on platelet reactivity, cigarette smoking increased collagen-induced platelet aggregation in healthy smokers 6 ; while another similar study did not observe differences in collagen-induced platelet aggregation following 8-hour abstinence from smoking. 18 In this study, the use of whole blood for platelet aggregation analyses overcomes limitations of previous studies that used platelet-rick plasma. This is important since the process of centrifugation in platelet preparation is thought to lead to activation of platelets and erroneous results. 19
The TXA2, produced by activated platelets during COX-mediated AA metabolism, is unstable and is rapidly hydrolyzed into the more stable and biologically inert TXB2. 20 Released in substantial amounts by activated platelets, TXB2 is further metabolized to 11-dhTXB2 and 2,3-dinor TXB2. 20 Urinary 11-dhTXB2 is considered an in vivo surrogate of TXA2 formation. 20 In this study, smokers had higher levels of TXB2 and 11-dhTXB2 in plasma and urine than nonsmokers, and cigarette smoking led to further increase in these levels acutely. This acute trend, however, was not observed in ex vivo production of TXB2 in platelets. This discrepant observation may be explained by depletion in the substrates required for TXB2 production ex vivo, thus preventing further increases in TXB2 when acutely stimulated after isolation. Our observations are consistent with those of the previous studies that measured TX levels. 21,22 A 4-year follow-up study involving 87 young male smokers found increased formation of TXA2 among chronic smokers, 21 while urinary TXA2 was higher in female smokers compared to nonsmokers. 22 In the latter study, urinary TXA2 levels corresponded closely with the frequency and number of cigarettes smoked. 22 Increased TXA2 biosyntheses were also reported in other studies that included chronic smokers, 9,10 where cigarette smoking was shown to facilitate the formation of TXA2 in platelets without affecting the overall survival of platelets in healthy females. 10 Similar to our findings, plasma concentrations of TXB2 increased immediately after cigarette smoking. 9 These studies, however, lacked correlation between the in vivo production of TX with marker of platelet aggregation. Data from the current study suggest that plasma TXB2 levels correlated closely with urinary TXB2 in chronic smokers and nonsmokers. The levels of TXB2, however, were higher in plasma (but not in urine) acutely after cigarette smoking, a finding that may be related to the half-life of TXB2 in plasma prior to its clearance in urine. These findings suggest that plasma TXB2 may be a sensitive marker of immediate changes in the in vivo production of TXA2 compared to urinary measurements.
Our findings suggest that upregulation of platelet COX-1 pathway may modulate the increase in platelet reactivity among chronic smokers. Several markers specific to this pathway (eg, TXB2 and platelet COX-1 expression) were increased in smokers compared with nonsmokers. Also, collagen-induced platelet aggregation associated significantly with platelet COX-1 activity (platelet TXB2 production) and COX-1 expression in smokers. Although previous studies had demonstrated that cigarette smoking increase the production of TXA2 in vivo, 9,10 the mechanisms underlying this rise are not known. Tobacco was previously shown to induce COX-1 expression in U937 human macrophages. 23 Despite their anucleate status, platelets are able to synthesize COX-1 de novo upon activation through a novel mechanism called signal-dependent pre-messenger RNA (mRNA) splicing. 24 The increased COX-1 platelet expression in smokers may, in part, offer an explanation for the observed aspirin resistance in this cohort. On the other hand, the lack of association between COX-1 expression, ex-vivo COX activity, and plasma TXB2 levels after acute smoking suggests that the further increase in TXA2 production after smoking is not explained by the rise in platelet COX activities. Although data from a meta-analysis of 6 primary prevention studies that include older smokers with established risk factors suggest a lack of efficacy of aspirin (a COX inhibitor) in preventing the development of cardiovascular events, 25 it is not known whether aspirin may be beneficial in young smokers without cardiovascular risk factors, in whom COX-1-mediated reactivity is significantly altered.
The MPV is closely related to cardiovascular risk factors such as cigarette smoking, 26 impaired fasting glucose, 27 hypertension, 28 hypercholesterolemia, 29 obesity, 30 and metabolic syndrome 29 and has been suggested as a marker of platelet activation. 30 In a study of 142 elderly smokers, the MPV levels were found to be significantly higher compared to nonsmokers, possibly from the stimulation of megakaryocytes to produce larger platelets. 26 In this study, peripheral platelets in smokers are larger than those in nonsmokers (mean MPV, 11.6 vs 10.8 fL). The significant association between collagen-induced platelet aggregation and platelet volume in this study suggests that MPV may reflect COX-1-mediated platelet reactivity. Following activation, platelets are capable of promoting atherosclerosis, by triggering vascular inflammation and thrombosis. 31 Data from the current and a previous study 7 suggest that young smokers are at risk of developing premature atherosclerosis through alterations in the platelet and endothelial functions.
These findings, however, are limited to young men smokers with no overt cardiovascular diseases. Information regarding the number of cigarette smoked in the week per month and 24 hours prior to the study enrollment was not systematically collected. Results from this study may be limited to light smokers (approximately 4 cigarette packs) and may not be reflective of the general smoking population. Despite the relative youth of study participants (mean age 29 years) and low cigarette exposure, our results showed that even low levels of cigarette smoking can bring about significant changes in COX-mediated platelet reactivity. It is not known whether heavy smokers or patients with established vascular risk factors would manifest similar changes in the platelet reactivity and aggregation profiles. Although smoking cessation remains the principal goal in primary prevention management strategies, the benefits of antiplatelet therapy to ameliorate the potential deleterious effects of platelet reactivity and aggregation among smokers remain unknown.
Footnotes
Authors’ Contributions
RCSS and WML conceived the idea of this work. RCSS, ECHL, and AMLQ recruited the study participants and collected the samples. WML, KLMS, CYJL, WLC, SEC, HH, and WFL performed the experiments. RCSS and WML performed the literature review and statistical analyses. RSCS and ECHL critically reviewed and finalized the manuscript. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the Singapore National Research Foundation and the National Medical Research Council.