
Abstract
This study was conducted on patients with a history of congenital bleeding disorders or with suspected bleeding tendencies. Laboratory analysis revealed Von Willebrand disease (VWD) in 68 (21.3%) of 318 participants with male to female ratio of 0.8: 1 (31 to 37) and median age 17 years (range 2-45 years). Type 3 being the most frequent, 35 (51.4%) of 68, type 2, 20 (29.4%) of 68, and lastly type 1, 13 (19.1%) of 68. A total of 55.8% patients with VWD presented with mucocutaneous bleeding. Menorrhagia was the most common presentation of female patients. Von Willebrand disease (21.3%) was the second common bleeding disorder and the most common coagulation defect among females with menorrhagia. However, the frequency in the study was quite low when compared to the western world. Similarly, low frequency of VWD type 1 might be due to the fact that only symptomatic patients visited us. Further studies are needed as there is limited information on VWD in the developing countries. This will help in the development of expertise for the accurate diagnosis & proper management.
Introduction
Von Willebrand disease (VWD) is a relatively common bleeding disorder caused by deficiency or dysfunction of Von Willebrand’s factor (VWF). It is associated with a defect in primary hemostasis and also with a secondary defect in coagulation factor VIII. 1 Clinically, it manifests typically as mucocutaneous type of bleeding and menorrhagia in female. Due to the high complexity of VWD, a whole panel of laboratory tests is required to diagnose and classify VWD, an essential step for the subsequent clinical management.
Worldwide VWD prevalence is generally 1% of the normal population with type 1 VWD being the most common, whereas its prevalence is 1.1% in United States.2,3 Von Willebrand disease is classified into partial quantitative deficiency (type 1) that accounts for 70% to 80% of all VWD, qualitative deficiency (type 2) that accounts for 20% VWD, and total deficiency (type 3) for approximately 5% to 10% resulting in secondary severe deficiency of FVIII. Type 2 VWD is divided further into 4 variants (2A, 2B, 2M, 2N) on the basis of the phenotype. 2 Although not many studies are available on the prevalence of VWD in Pakistan, some of the tertiary care centers and institutions have reported a prevalence of 3.18% to 7.74% among the cases with hereditary bleeding disorders. 4 Von Willebrand disease therefore remains an underdiagnosed or misdiagnosed entity in Pakistan and the prevalence of different subtypes of VWD is also not known. The present study was carried out to know the frequency of VWD, its subtypes, and clinical features among the patients seeking advice for bleeding tendencies.
Patients and Methods
This study was conducted at National Institute of Blood Diseases from August 2007 to March 2009. It was approved by hospital ethics committee and was carried out in accordance with declaration of Helsinki. This was a cross-sectional, multicenter epidemiological study. Informed consent was obtained from all adult participants, parents, or legal guardians. Patients with a history of congenital bleeding disorders or suspected bleeding tendencies were screened. Patients known to have acquired VWD, immune thrombocytopenic purpura (ITP), thrombotic thrombocytopenic purpura (TTP), disseminated intravascular coagulation (DIC), or on anticoagulant therapy were excluded. A washout period of 7 days was set before blood samples were taken for analysis. A detailed personal and family history of bleeding manifestations including onset, signs, and symptoms were noted on the study performa. Complete Blood Count (CBC), bleeding time (BT), Prothrombin time (PT), Activated Partial Thromboplastin Time (APTT), fibrinogen and blood grouping were the laboratory tests performed on the same day for the patients who met inclusion criteria. For coagulation assays, venous blood samples were collected in tubes containing 0.109 M (3.2%) trisodium citrate in a ratio of 9 parts blood to 1 part anticoagulant and then centrifuged without delay at 1200 G for 15 minutes. 5 The remaining plasma was stored in 2 aliquots at −70°C for factor assay, VWD diagnosis, and subtypes.
Blood sample for CBC collected in EDTA tubes was performed on Sysmex XE-2100 (Kobe, Japan). PT, APTT, factor VIII levels, and VWF: Ag was performed on Sysmex CA-1500 (Kobe, Japan) using appropriate quality control materials and standard reagents (Dade Behring, Germany). PT, APTT, and BT were performed using standard techniques 5 while factor assays were done by one-stage coagulometric method (factor VIII normal range: 50%-150%). Clauss method was used for fibrinogen assay and BT was performed by Ivy’s modified template method. 5 Von Willebrand’s factor: Ag was analyzed using Diagnostica Stago reagent, calibrated on Sysmex CA-1500; test was based on immunoturbidimetric method (normal range: 50%-160%) while Ristocetin cofactor assay (RiCoF) was carried out using stabilized platelets agglutinated in the presence of VWF and the antibiotic ristocetin on AggRAM (manufactured for Helena BioSciences Europe) normal range >57%). Ristocetin-induced platelet aggregation (RIPA) was done on AggRam (Helena Laboratories) using platelet rich plasma (PRP) with a count adjusted to 200 to 250 × 109/L. 6 Local reference ranges were calculated for these tests in our laboratory on healthy male and female individuals, n = 80. Von Willebrand disease is classified on the basis of criteria developed by the VWF Subcommittee of the International Society on Thrombosis and Haemostasis (ISTH), first published in 1994 and revised in 2006.7–9 The RiCoF to VWF: Ag ratio of <0.7 is used in differentiating type 1 from type 2 VWD. 10 Type 3 VWD is characterized by undetectable to less than 10% VWF protein with markedly decreased activity and very low FVIII levels (1-9 IU/dL). 11
Statistical packages for social science (SPSS-13) were used to analyze data. Frequency and percentage were computed for categorical variables and mean and standard deviation were estimated for quantitative variables.
Results
Three hundred and seventy six patients were evaluated for inherited bleeding disorders in our centre. Hemostatic abnormalities were found in 318 patients. Among them were hemophilia A, hemophilia B, VWD, and platelet functional disorders. Von Willebrand disease was found in 68 (21.3%) of 318 participants with male to female ratio of 0.8:1 (31/ 37) and median age 17 years (range 2-45 years). Of the 68 (72.0%) patients, 49 gave a positive family history of bleeding tendencies. Type 3 was the most frequent, 35 (51.4%) of 68, type 2, 20 (29.4%) of 68, and lastly, type 1, 13 (19.1%) of 68. 55.8% VWD patients presented with mucocutaneous bleeds such as gum bleeding, epistaxis, bruises, etc. Menorrhagia was the most common presentation among female patients with VWD. Median age of female patients with menorrhagia was 15.2 years. All of them had hypochromic microcytic anemia, Hb mean 7.6g/dl and were treated with iron supplements, oral contraceptives (Combination of estrogen–progesterone), antifibrinolytic agents (tranexamic acid), alone or in combination. Desmopressin, a synthetic analogue of vasopressin (0.3 mg/kg SQ (1–3 doses) and intranasal 300 mg/ day for2-3 days was used only in 4 patients of type 1 VWD. The need for blood-product replacement i.e. fresh frozen plasma and cryoprecipitate occurred in 82% of women with type 2 and 3 VWD unresponsive to other treatment .Pack red cells transfusion was given to six patients of Hb less than 6.0 g/dl with signs of anemia.
In type 3, presentation was similar to patients with hemophilia having hemarthrosis (19.1%) and hematomas (30.9%), details given in the Table 1 . We also had 11 patients with low VWF levels, levels ranging from 30.92% ±2.99 below the normal reference range.
Clinical Spectrum of Von Willebrand Disease a
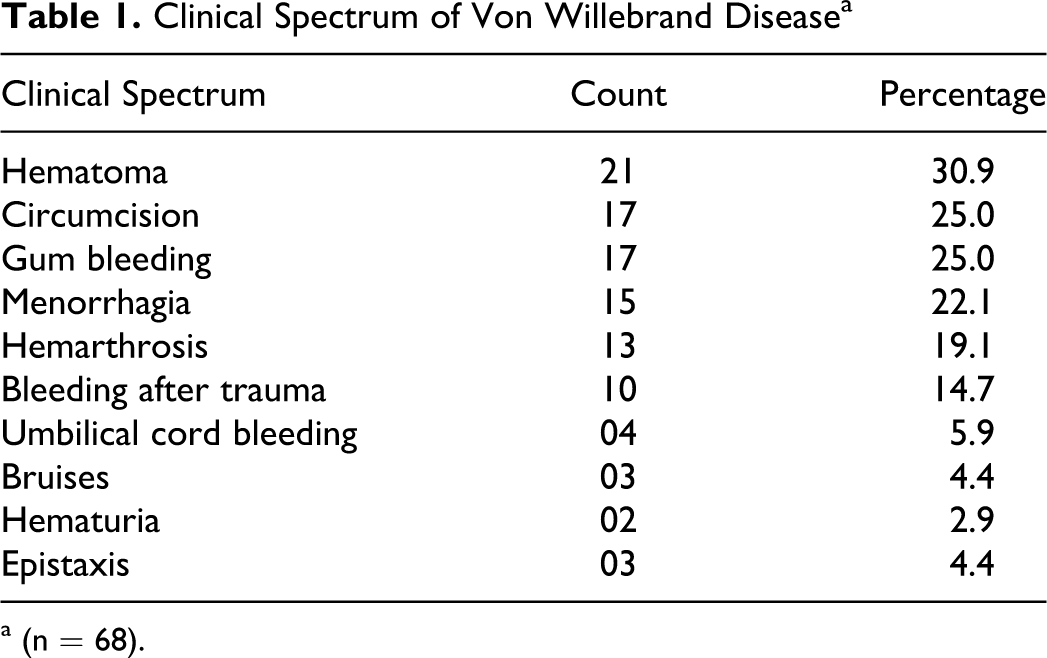
a (n = 68).
Laboratory Parameter and Blood Groups With Respect to Type of VWD
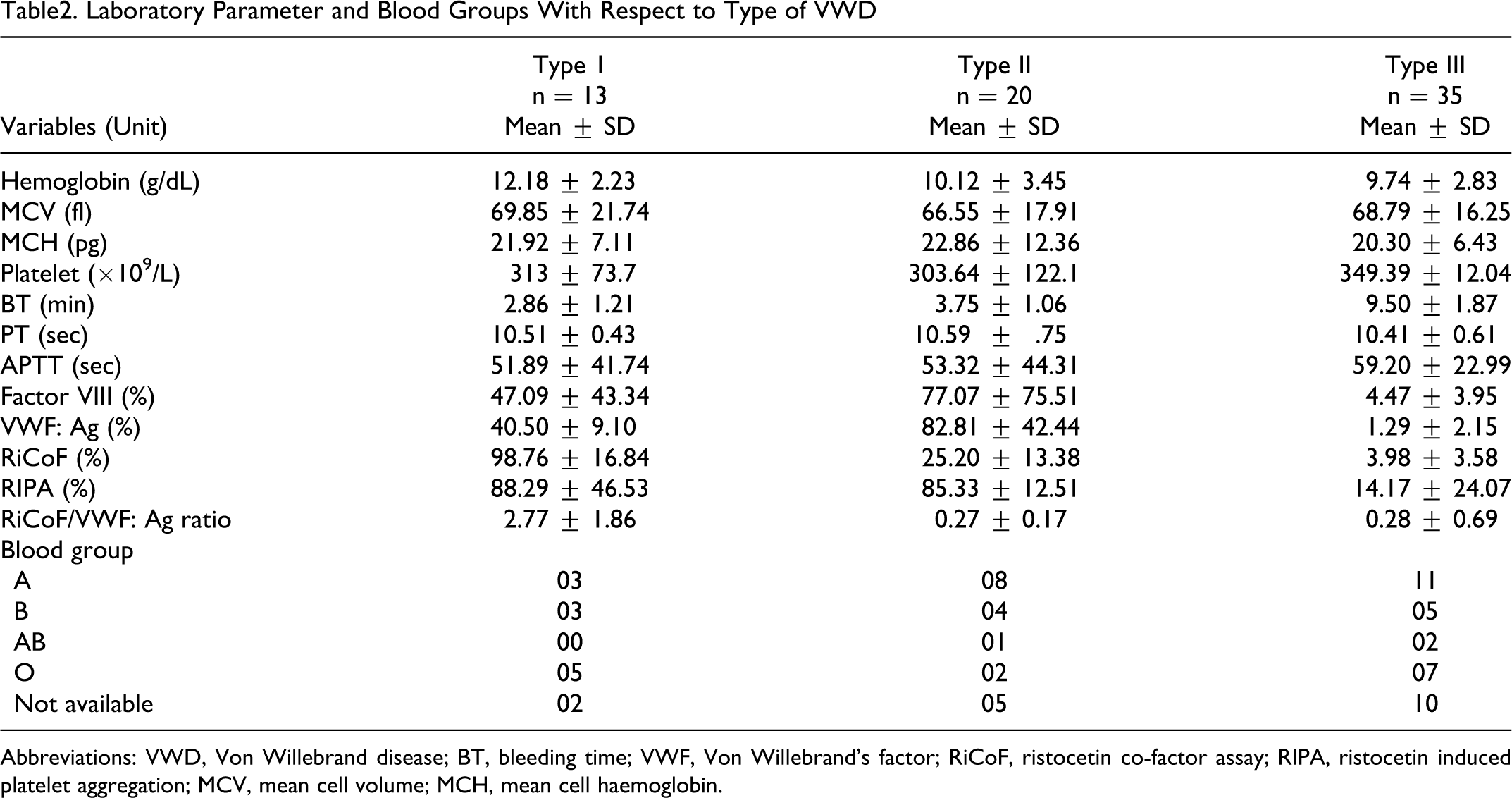
Abbreviations: VWD, Von Willebrand disease; BT, bleeding time; VWF, Von Willebrand’s factor; RiCoF, ristocetin co-factor assay; RIPA, ristocetin induced platelet aggregation; MCV, mean cell volume; MCH, mean cell haemoglobin.
Discussion
The incidence of congenital bleeding disorders may vary according to country and ethnic origin. Studies indicate that VWD is the most common inherited bleeding disorder in the western world.1,2 But studies conducted from countries like India and Iran showed low prevalence of VWD.12–14 Our data showed 21.3% (68 of 318) frequency of VWD among patients with positive family history of bleeding in first degree relatives. Interestingly, type 3 VWD was the most common, seen in 51.4% (35 of 68) of patients, type 2 in 29.4% (20 of 68), and type 1, in 19.1% (13 of 68). The diagnosis of higher number of patients of type 3 VWD could be due to the fact that these patients were most severely affected and symptomatic; on the other hand, patients with type 1 VWD had mild symptoms, as a result lesser number sought for medical advice, leading to underdiagnosis of milder type 1 VWD. Type 1 VWD was seen in 19.1% patients in contrast to 60% to 70% prevalence of type 1 VWD reported in studies from other countries.1,2,15 The patients with type 1 VWD have mild bleeding episodes and hence consider their bleeding tendency as normal, unless they come across a major hemostatic challenge like trauma or surgery for which they get themselves investigated. These milder forms are often missed out on diagnosis or may not be detected at all as screening coagulation tests may be in the normal range.
Our study showed wide spectrum of clinical features of bleeding in our participants. Common manifestation included mucocutaneous bleeds (55.8%), with gum bleeding, epistaxis, menorrhagia, and bruises being common among these patients. It was interesting to note that the symptoms varied among affected members of the same family and also over time in a single individual. Menorrhagia (22.1%) was the most common presentation of female patients with VWD in our study. Menorrhagia is a valuable predictor of a bleeding disorder in women mostly beginning at menarche. The frequency of VWD in women with menorrhagia ranges from 5% to 20% in different studies16,17 and often it is the only symptom in mild type 1 disease.18–20 The data from multiple studies strongly suggested that VWD was more prevalent in women with menorrhagia than in the general population. 20 Therefore, the specificity of menorrhagia as a predictor of VWD can be estimated as 5% to 20%. All patients with VWD were treated with factor replacement therapy either with fresh frozen plasma or cryoprecipitate. Plasma-derived viral-inactivated or recombinant factor concentrates were used only in 15% of cases, provided by World Federation of Haemophilia (WFH) or other donor agencies.
We also had 11 patients with low VWF levels (30.92% ± 2.99%) below the normal reference range. This category of patients, having low levels of VWF verses type 1 has been documented in the literature particularly in association with blood group “O.”1,2 Persons with very low VWF levels, <20 IU/dL, are likely to have VWF gene mutations, significant bleeding symptoms, and a strongly positive family history. 21 But VWF levels of 30-50 IU/dL, just below the usual normal range (50–200 IU/dL), pose problems for diagnosis and treatment.
Conclusion
Von Willebrand disease (21.3%) was the second common bleeding disorder and most common coagulation defect among females with menorrhagia. However, the frequency in the study is quite low when compared to the western world. Similarly, low frequency of VWD type 1 might be due to the fact that only symptomatic patients had visited us. A total of 50% male patients were previously labeled as hemophilia A. Further studies are needed as there is limited information on VWD in the developing countries. For the accurate diagnosis of bleeding disorders, establishment of coagulation laboratories with proper training of technical staff is the need of time, which in turn will help in better patient care.
Footnotes
Acknowledgment
We would like to thank Novo Nordisk Haemophilia Foundation for their financial and technical support for Capacity Building of Haemophilia Care in Karachi region. Without their generous support, this study could not have been possible.
The author(s) declared no conflicts of interest with respect to the authorship and/or publication of this article.
Dr. Tahir Shamsi (Medical Director) has received grant from NovoNordisk Haemophilia Foundation.