
Abstract
Objective: To determine the frequency of inherited bleeding disorders, its complications, and treatment modalities available for its treatment. Design: Cross-sectional study. Patients and Methods: Patients with a history of bleeding tendency were tested for confirmation of the diagnosis. History and clinical findings were recorded. Laboratory analysis included prothrombin time (PT), activated partial thromboplastin time (APTT), bleeding time (BT), and fibrinogen assay. Patients with prolonged APTT were tested for factors VIII (FVIII) and IX (FIX). If FVIII was low, von Willebrand factor: antigen (vWF:Ag) and von Willebrand factor:ristocetin cofactor (vWF:RCo) were performed. When PT and APTT both were prolonged, FV, FX, and FII were tested. Platelet aggregation studies were done when there was isolated prolonged BT. Urea clot solubility test was done when all coagulation tests were normal. All patients with hemophilia A and B were evaluated for inhibitors. Results: Of the 376 patients, inherited bleeding disorder was diagnosed in 318 (85%) cases. Median age of patients was 16.4years. Hemophilia A was the commonest inherited bleeding disorder that was observed in 140 (37.2%) followed by vWD 68 (18.0%), platelet function disorders 48 (12.8%), and hemophilia B in 33 (8.8%) cases. We also found rare congenital factor deficiencies in 13 (3.4%), low VWF in 11 (3.0%) participants and 5 (1.3%) in female hemophilia carriers. Hemarthrosis was the most frequent symptom in hemophilia A and B (79.7%) involving knee joint. Inhibitor was detected in 21 (15%) cases. Fresh frozen plasma/cryoprecipitate were the most common modality of treatment. In 58 patients, no abnormality was detected in coagulation profile. Conclusion: Hemophilia A and vWD are the most common congenital bleeding disorders in this study. Hemarthrosis involving knee joint was the most common complication. Inhibitor was detected in a significant number of patients. Plasma is still the most common modality of treatment.
Keywords
Introduction
The bleeding disorders are known to treating physicians since 16th century. 1 Medical literature in the West is full of knowledge regarding different aspects of these disorders. Congenital bleeding disorders are found in all racial groups and have worldwide distribution but very limited information is available in developing countries like Pakistan about their prevalence and so on. Deficiencies of congenital coagulation factors result in patients with bleeding disorders. Among all, hemophilia A (HA), B (HB), and von Willebrand disease (vWD) are the commonest and are characterized by low levels of factor VIII(FVIII)/IX (FIX) or von Willebrand factor (vWF), respectively. 1 Severity of bleeding is proportional to the severity of factor deficiency; therefore, HA and HB diseases, X-linked recessive disorders, are classified as mild, moderate, and severe according to concentration of factor present in the plasma.2,3 Rare bleeding disorders are mainly autosomal recessive and result from deficiencies of clotting factors, other than factors VIII, IX, and vWD, or due to poor platelet functions. Higher incidence of platelet functional disorders has been reported from Middle East and India. 4 The incidence 5 of HA (FVIII deficiency) is 1 in 5000 live male births and that of HB (FIX deficiency) is 1 in 30 000. In contrast, the deficiency or dysfunction of vWF constitutes the most common bleeding disorder in females, the incidence of which may reach 1 in 1000 or even more. 6 In 30% patients, new mutation in FVIII gene results in hemophilia. 2
Clinically recurrent, spontaneous, and posttraumatic hemorrhages involving deep muscles, resulting in hematoma formation, hemarthrosis, and easy bruising are the characteristic features in patients with hemophilia. Infants may have excessive postcircumcision bleeding. Recurrent joint and muscle bleeds ultimately lead to crippling joint deformities and muscle wasting. At present no cure, for these diseases, is available. Clotting factor replacement therapy is the main stay in management of these patients. However, factor replacement therapy, like fresh frozen plasma (FFP), exposes the patient to increased risk of transfusion transmitted diseases for example HCV, HBV, and HIV infections and also end up in alloantibodies (inhibitor) formation against the missing factor. These antibodies neutralize the procoagulant function of therapeutically administered factor and ultimately make the patient resistant to conventional factor replacement therapy. Inhibitors develop with frequency of 20% to 30% in HA and 02% to 5% in HB.7,8
In Pakistan, hemophilia management is not provided by the government. There is no infrastructure available to look after this lifelong bleeding disorder. Laboratory diagnosis of hemophilia is limited to teaching hospital in the bigger cities like Karachi. Most patients get FFP or occasionally cryoprecipitate in the public sector hospitals in the bigger cities and only patients, who are registered with hemophilia societies, get appropriate treatments. Virally inactivated high-purity or intermediate purity factor concentrates are out of the reach for the majority of patients. In the smaller cities, towns, and villages they are not even diagnosed. Although small public hospitals, basic health units, and dispensaries run by the health department do exist in the smaller cities and villages, they do not even get the basic management and rather are mismanaged due to lack of awareness of the disease by the patients and health providers, thus leading to high morbidity and mortality among these patients. With this background, the study was designed with the help of Novo Nordisk Haemophilia foundation (NNHF) to determine the frequency of bleeding disorders in Karachi region and assess the presenting features, extent of complications, and therapeutic options available to these patients. The study was carried out with the secondary aim to update the knowledge and skills in the diagnosis of bleeding disorders and prevention of complications related to the disease and its treatment.
Material and Methods
This study was conducted from August 2007 to March 2009. The study was approved by institutional ethics committee and was done in accordance with the declaration of Helsinki. Informed consent was obtained from all adult participants, parents, or legal guardians. This was a cross-sectional, multicenter epidemiological study. Patients from all hemophilia treatment centers, who either have a personal and family history of congenital bleeding disorders or a suspected bleeding tendency were screened through free clinics and free laboratory diagnoses. Known patients of immune thrombocytopenic purpura (ITP), thrombotic thrombocytopenic purpura (TTP), disseminated intravascular coagulation (DIC), or patients on anticoagulant therapy were excluded. A washout period of 7 days was set before blood samples were taken for analysis. A detailed history including symptoms and signs at present and at the time of onset of disease, family history, and clinical examination was recorded on the study performa. Type and site of bleeding, presence or absence of joint deformity, number of joint involved, hematoma formation, intracranial bleeds, duration, and type of therapy including surgical intervention used were also recorded. Baseline laboratory tests including complete blood count (CBC), bleeding time (BT), prothrombin time (PT), activated partial thromboplastin time (APTT), fibrinogen, and blood grouping were carried out in all patients on same day. For coagulation assays, venous blood samples were collected in tubes containing 0.109 mol/L (3.2%) trisodium citrate in a ratio of 9 parts blood to 1 part anticoagulant and then centrifuged without delay at 1200g for 15 minutes. The remaining plasma was stored in 2 aliquots at −70°C for factor assay, vWD diagnosis, and inhibitor screening in batches. Serum was used for Serum glutamic pyruvic transaminase (SGPT), hepatitis B, hepatitis C, and HIV tests.
CBC was done on XE-2100 hematology analyzer (Sysmex, Kobe, Japan). Coagulation profile was done on CA-1500 (Sysmex), using appropriate quality control materials and standard reagents (Dade Behring, Germany). Prothrombin time, APTT, and BT were performed using standard technique, while factor assays were done by one-stage coagulometric method. 9 The measuring range for the determination of coagulation factors (VIII, IX, XI, and XII) extends from 1% to approximately 100% of the normal and can be increased to approximately 200% using a higher dilution of sample. Severity of hemophilia was labeled as severe, moderate, and mild according to factor levels of <1%, 1% to 5%, and >5%, respectively. 2 vWF:Ag was analyzed using Diagnostica Stago (France) reagent and calibrated on Sysmex CA-1500; test was based on immunoturbidimetric method with (normal reference range: 50%-160%). Ristocetin cofactor assay (vWF:RCo) was carried out using stabilized platelets agglutinated in the presence of vWF and the antibiotic ristocetin (normal range: >57%; AggRAM Helena Laboratories, United Kingdom). von Willebrand disease is classified into 3 major categories: partial quantitative deficiency (type 1), qualitative deficiency (type 2), and total deficiency (type 3). The classification was based on the criteria developed by the vWF Subcommittee of the International Society on Thrombosis and Haemostasis (ISTH), first published in 1994 and revised in 2006.10,11 The vWF:RCo to vWF:Ag ratio of <0.7 was used in differentiating type 1 from type 2 vWD. 12 However, other test like multimer analysis was not done due to nonavailability of kits. Type 3 vWD is characterized by undetectable vWF protein and activity, and FVIII levels are usually very low (1-9 IU/dL). 13
Clauss method was used for fibrinogen assay and BT was assessed by Ivy modified template method. 9 Hepatitis B, C, and HIV were done on Evolis (Biorad, France) based on enzyme-linked immunosorbent assay (ELISA). Blood groups were determined manually using tube method. All patients with HA and HB were evaluated for inhibitors. Inhibitors against FVIII were screened using APTT-based method, 14 50:50 patient’s plasma mixed with normal pooled plasma incubated together for 2 hours at 37°C. Factor IX inhibitors 15 were tested after 15 minutes incubation at 37°C. The inhibitors were quantified by Bethesda assay. Factor VIII inhibitors 9 are time dependent and are called low titer when less than 5 Bethesda unit (BU) detected while high titer means more than 5 BU.
Platelet function defect (PFD) was considered when BT was greater than 7 minutes with normal to low platelet count. Platelet aggregation studies were done on Helena AggRAM (Helena Laboratories) using ristocetin, adenosine diphosphate (ADP), epinephrine, and collagen. Platelet-rich plasma (PRP) was used with a count adjusted to 200 to 250 × 10 9 /L. Factor XIII screening was done by urea clot solubility test in patients where all coagulation tests were normal. 9
Results
A total of 376 patients were screened. Mean age was 16.4 years (range: 1month-60 years). Diagnosis was established in 318 patients, while in 58 patients, no abnormality in coagulation tests was found. Only 57 patients enrolled in the study knew their diagnosis before hand but in 24 patients, previous diagnosis was found incorrect. Family history of bleeding was present in 77.1% patients. Table 1 shows the demographic detail of patients in the study. Of 318, HA, diagnosed in 140 (37.2%) patients, was the most common bleeding disorder followed by vWD in 68 (18.0%), HB in 33 (8.8%), PFD in 48 (12.8%), and other rare inherited coagulation disorders (RICDs) in 13 (3.4%). The ratio of HA:HB was 4.2:1. Majority of hemophiliacs had moderate severity (52.0%) both in HA and HB; but as mentioned earlier, the kit used of Dade Behring for FVIII and FIX extends from 1% to approximately 100% of the normal (Figure 1). Inhibitors were detected in 21 patients with HA (<5 BU in 8 patients and >5 BU in 13 patients) while none in HB. Delayed presentation and diagnosis was a common feature. Figure 2 shows the life expectancy in patients with hemophilia as compare to normal population. 16 The most common coagulation defect in females was vWD. Hepatitis C antibodies were detected in 107 (28.4%) of 376, HBsAg was detected in 7 (1.8%) of 376, and HIV 1 and 2 antibodies was found in 1 (0.26%) of 376.
Demographic Data of Inherited Disease Patients
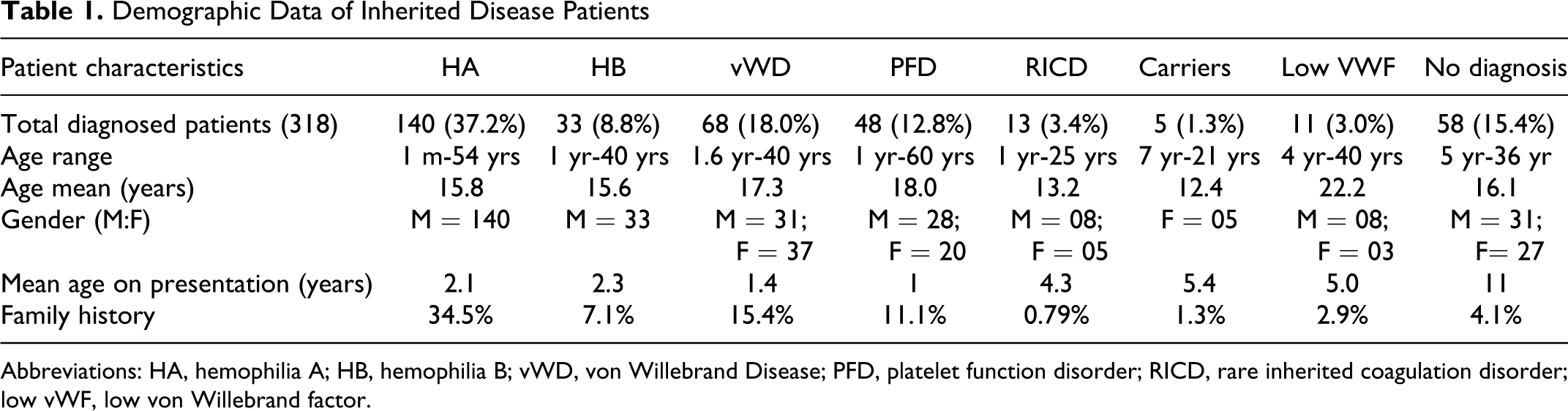
Abbreviations: HA, hemophilia A; HB, hemophilia B; vWD, von Willebrand Disease; PFD, platelet function disorder; RICD, rare inherited coagulation disorder; low vWF, low von Willebrand factor.
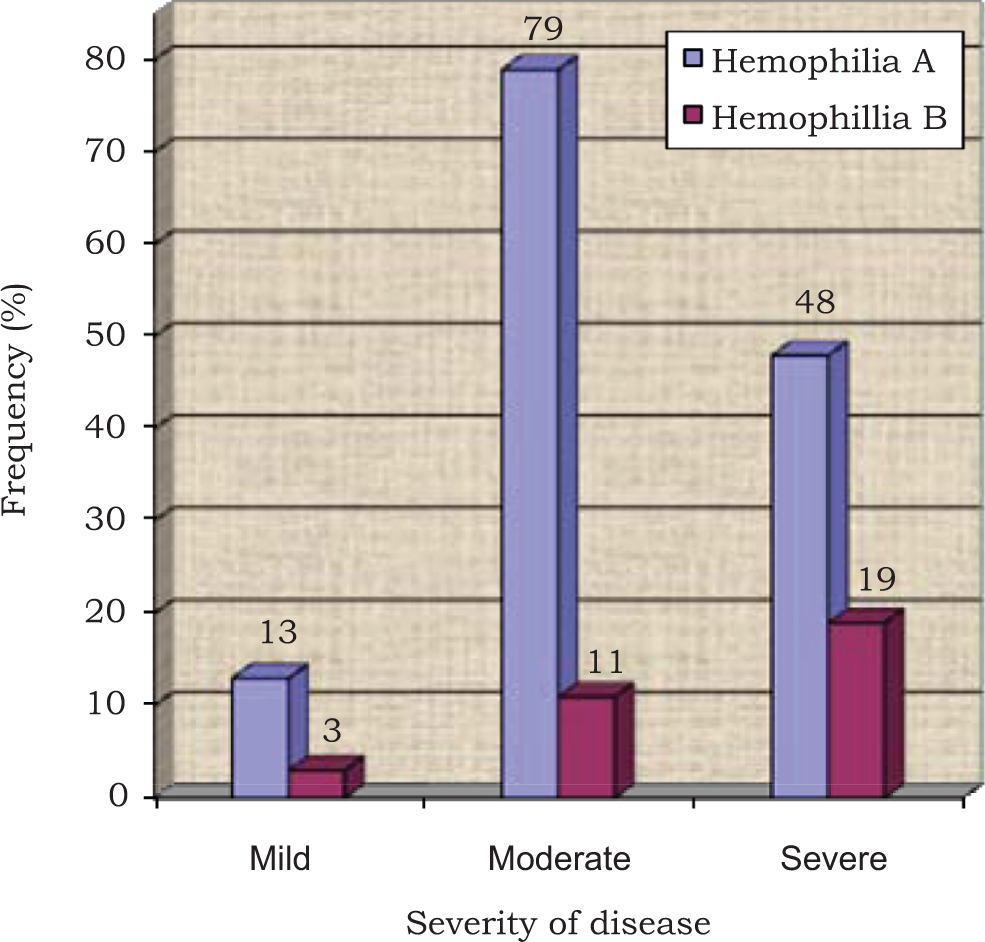
Distribution of hemophilia A and B according to severity (n = 173).
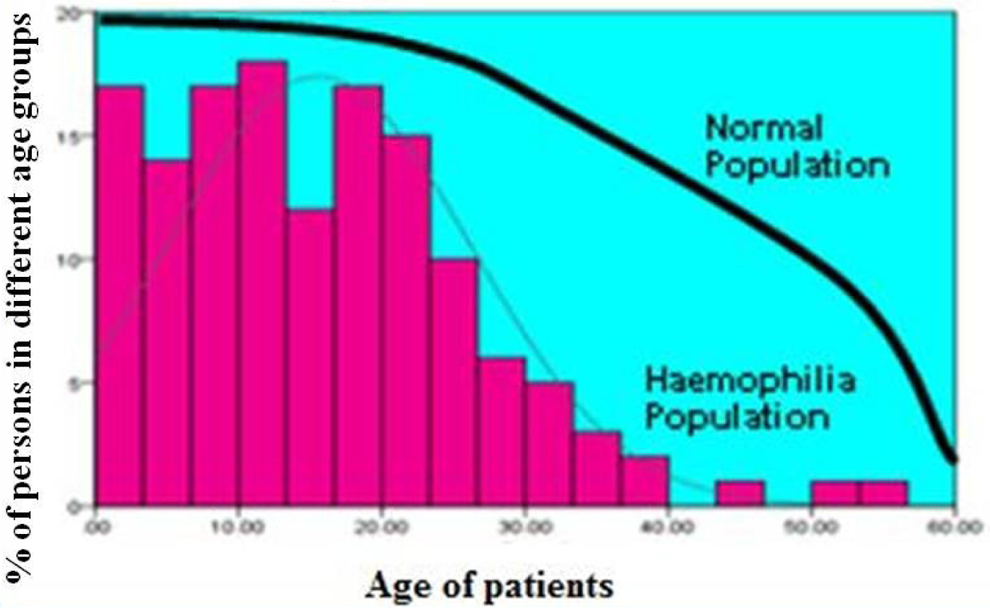
Life expectany of Patients with hemophilia.
Table 2 shows clinical manifestations of 318 diagnosed patients. The most common type of bleeding was hemarthrosis 138 (43.3%) in HA and HB; followed by hematoma 120 (37.7%), gum bleeding 43 (13.5%), hematuria 37 (11.6%), easy bruising 29 (9.1%), menorrhagia 21 (6.6%), intracranial hemorrhage (ICH) in 16 (5.0%), epistaxis 12 (3.7%) umbilical cord bleeding 8 (2.5%), malena in 3 (0.9%), gastrointestinal bleeding 3 (0.9%), while traumatic bleeding episodes were 67 (21.0%). Overall mucocutaneous bleeding was seen in 55.8% patients with vWD. Menorrhagia was the most common presentation among the female patients with vWD.
Clinical Spectrum of Bleeding in Congenital Bleeding Disorders
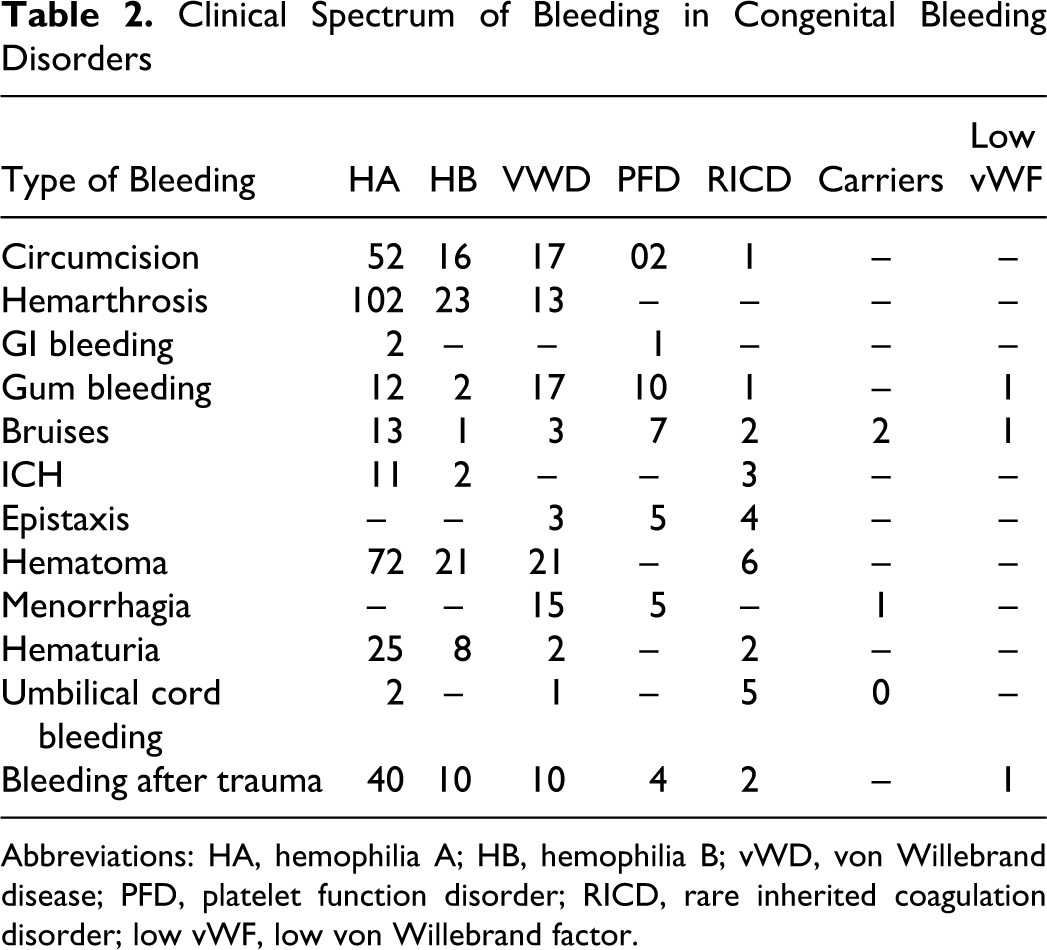
Abbreviations: HA, hemophilia A; HB, hemophilia B; vWD, von Willebrand disease; PFD, platelet function disorder; RICD, rare inherited coagulation disorder; low vWF, low von Willebrand factor.
Of platelet function disorders, Glanzmann thrombasthenia was diagnosed in 12 (3.19%), Bernard-Soulier in 4 (1.0 %), ADP receptor defect in 11 (2.9%), collagen receptor defect in 3 (0.79%), and epinephrine receptor defect in 11 (2.9%). In 7 (1.8%) patients, the PFDs could not be classified into specific group. The majority had a delayed diagnosis, although they had a history of bleeding manifestations since childhood. The mucocutaneous type of bleeding was the commonest (56.2%).
In all, 13 (4.0%) patients with rare bleeding disorder. Factor VII deficiency was detected in 7 (1.8%) cases, FX deficiency in 2 (0.53%) afibrinogenemia in 2 (0.53%), while FV deficiency in 1 (0.26%) and FXIII deficiency in 1 case (0.26%). There were 2 cases of combined factors V and VIII deficiency (0.53%), and both of these patients had ICH due to severe deficiencies. In these patients with mucosal hemorrhage (69.2%), gum bleeding, epistaxis, easy bruising, and bleeding associated with trauma were the presenting feature, but none of them had hemarthrosis. History of umbilical cord bleeding was present in only patients with FXIII deficiency and 1 patient each with afibrinogenemia and FVII deficiency.
Eleven participants were identified with low VWF:Ag levels (37.92% ± 4.99%) below the normal reference ranges established with different blood groups. We were also able to identify 5 HA carriers (FVIII 28.3% ± 3.34%). Among them 1 female presented with menorrhagia and others with history of easy bruising. All had positive family history of bleeding.
Treatment records of all these patients are shown in Table 3 . Fresh frozen plasma, cryoprecipitate, and its components were used most frequently; viral inactivated factor concentrate (plasma derived or recombinant) were used when available.
Treatment Given to Patients
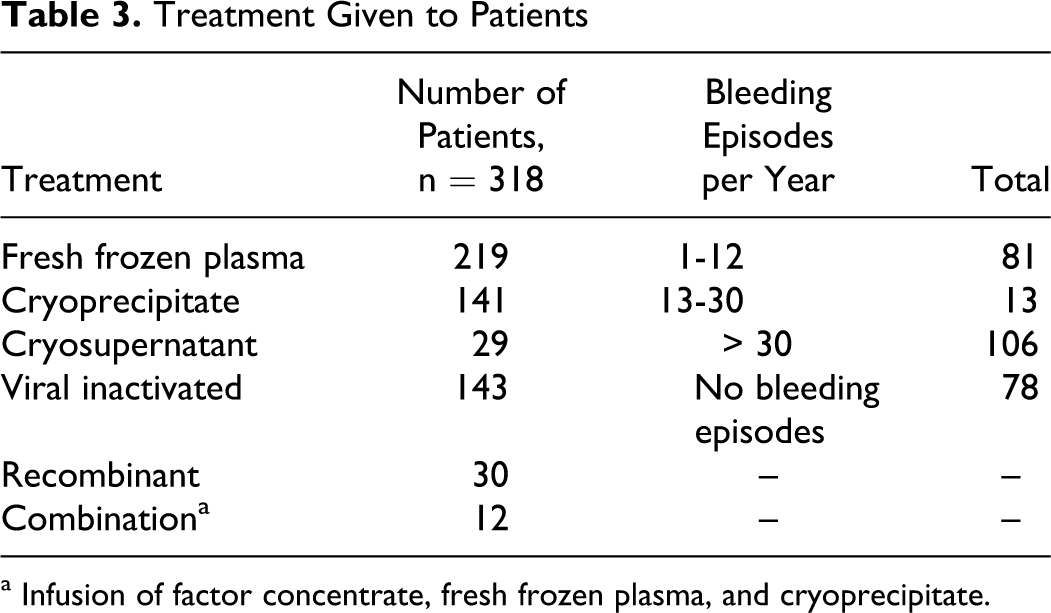
a Infusion of factor concentrate, fresh frozen plasma, and cryoprecipitate.
Discussion
As about 80% of the total number patients with hemophilica live in the developing world, and most of these people live where they do not have access to diagnosis and adequate treatment. 17 But most of the epidemiological data on bleeding disorders is compiled in the West or developed countries. Karachi is the largest city of the country, with a population of about 16 million; almost all ethnic groups from various areas of the country have representation in this city. The objectives of this study were to determine the frequency and spectrum of bleeding disorders in Karachi region. Secondary goals of the study was, on the basis of the collected data, to develop and improve diagnostic facilities, provide better care, and in particular increase awareness among masses, involve government in hemophilia care, and educate patients and their families and health care community.
The number of persons with hemophilia is about 400 000 worldwide, which affects males of all ethnic groups. Globally, HA is more common than HB.17,18 Literature search shows that vWD is the most common hereditary bleeding disorder in the developed world.11,12 Platelet function defect are relatively rare in these countries. A relatively high incidence of autosomal bleeding disorders and platelet function abnormalities has been reported from the Middle East and also from India. 19
Our study has shown HA as the most common bleeding disorder with decreased life expectancy when different age groups of patients with hemophiliac a are compared with normal population (Figure 2). Earlier study from Pakistan by Zafar et al has shown similar findings. 7 These figures are close to the figures found in neighboring countries such as Iran and India.20,21 Karimi et al have reported 367 patients with inherited coagulation disorders from Iran. A total of 272 (73.8%) patients had HA, 39 (10.6%) had HB, and 24 (6.5%) were diagnosed with vWD. The incidence of PFDs was, however, not reported in this study. 20 Similarly studies published from India also illustrated that HA being the most common inherited bleeding disorder, with the variable ratio ranging from 8 to 4:1 for HB. This is closely followed by PFD in India, which are quite rare in the industrialized world. von Willebrand disease was found to be relatively rare in the Indian population. 21 However, in our series vWD was found in 68 (18.0%) patients and PFD in 48 (12.8%). We were also able to identify 5 hemophilia carriers (FVIII 28.3% ± 3.34%) with positive family history of bleeding in first-degree relatives. Majority were asymptomatic, while 1 had menorrhagia and 2 females complained of easy bruising. Carriers are usually asymptomatic but a few carriers may have clotting factor levels in the hemophilia range, mostly in the mild category, but in rare instances, carriers can be in the moderate or severe range due to extreme lyonization or due to marriage of a carrier to an affected male, usually a cousin. 1
Due to the lack of proper knowledge and diagnostic facilities even in large cities of Pakistan, a significant number of patients with bleeding disorders either are not diagnosed or miss diagnosed as evident in this study, 24 patients did not have proper diagnosis. In all, 58 (15.4%) patients showed normal parameters when tested for bleeding disorders. We did not perform prothrombin consumption index (PCI) neither screened the patients for defects of fibrinolytic system. A study from India has shown 19% patients had isolated platelet factor 3 availability defect. 21 On the other hand, fibrinolytic system defect usually present with thrombotic complications, but bleeding manifestations have also been reported in this subset of patients. 22
Clinically HA is indistinguishable from HB. Patients with severe hemophilia experience frequent spontaneous bleeding episodes, in contrast to those with moderate and mild hemophilia in whom trauma or surgery usually provokes hemorrhage. Most of our patients with HA and HB presented with bleeding after circumcision or hemarthrosis (intra-articular bleeding), which is the most common clinical manifestation involving ankle, knee, and elbow joints most frequently. In our series, 43.3% patients had hemarthrosis involving knee, ankle, elbow, and wrist joints.
Current treatment for hemophilia-related bleeding comprises plasma-derived or recombinant factor concentrates. This treatment is expensive, restricts the prophylactic use of factors that can lead to crippling joint disease, and may transmit infectious agents. But more than 80% of the patients with hemophilia live in countries with few medical or financial resources. 17 Fresh frozen plasma remains the most commonly used therapeutic modality in our patients due to either nonavailability or high cost of factor concentrates. Blood products are derived in our blood bank from mostly exchange blood donors. Studies conducted in Pakistan on blood donors have shown prevalence of various infectious pathogens that are ranged as follows: hepatitis B 1.46% to 2.99%, with a downward trend over time, hepatitis C 3.01% to 4.99%, HIV 0% to 0.06%, respectively.23,24 For HIV, a prevalence rate of 0.1% among the general population has been reported by the World Bank, but lack of awareness in Pakistan, cultural issues, and high-risk factors including low condom use and unscreened use of blood make the situation fertile for AIDS to become a major health issue. 25
In the present study, only 45% were treated with viral inactivated factor concentrates provided by World Federation of Hemophilia (WFH) or other donor agencies. Replacement therapy with factor concentrate may result in HCV, hepatitis B, and HIV infection and also development of inhibitors. Though virus inactivation methods have virtually eliminated the transmission of these viruses in newer factor concentrates, however the risk remains in developing countries where there is no ready access to these concentrates. 26 Partially screened blood products have taken their toll as significant number of patients is infected with HCV (28.4%), but low HBV (1.8%) depicts overall downward trend HBV infection rate. Therefore, transfusion transmitted infections continue to have a significant impact on patient management.26,27 Plasma or other blood products are not always screened for transmissible agents. Suboptimal factor replacement, lack of education about simple technique for example application of ice or ice packs, immobilization of the affected joint, use of slings and so on, lack of physiotherapy facilities contribute to the physical disability and crippling arthropathy in these patients.
Inhibitors are antibodies that neutralize FVIII activity and develop in 20% to 30% of patients with severe and 5% to 15% of patients with mild-to-moderate HA. 28 The inhibitor development in HB is relatively uncommon occurring in 2% to 3% of patients with HB. 28 It has been shown in studies that large deletions, stop codon mutations, and inversion 22 associated with greater chances to produce inhibitor as compared to small deletions and missense mutations (35% vs 5%). Also, exposure to high-purity products early in life and family history of inhibitors increase the risk of its development. 29 An important observation in our study was development of inhibitor in 21 (15%) patients with HA, with 8 participants having <5 BU titer, while 13 have >5 BU titer. Screening tests for inhibitors were negative in patients with HB. Presence of relatively modest number of inhibitors in patients with hemophilia could be due to the more frequent use of FFP or cryoprecipitate than purified products (Table 3). However, we were unable to identify the risk factors for inhibitor development in these patients.
von Willebrand disease is relatively a common autosomal inherited bleeding disorder caused by deficiency or dysfunction of vWF which mediates the initial adhesion of platelets at sites of vascular injury and also binds and stabilizes blood clotting FVIII in the circulation. 10 The prevalence30,31 is about 23 to 110 per million population (0.0023%-0.01%)). Common manifestations include mucocutaneous bleeding, menorrhagia, gum bleeding, epistaxis, bleeding after dental extraction, postoperative bleeding, and rarely hemarthrosis. 30 In this study, vWD is found as the second most common inherited bleeding disorder 68 (21.3%), type 3 being most frequent 35 (51.4%), followed by type 2, 20 (29.4%) and type 1, 13 (19.1%). The low frequency of type 1 vWD in this study could be due to lack or mild nature bleeding manifestations in type-1 VWD disease. We also had 11 patients with low vWF levels (37.92 ± 4.99), below the normal reference range. The association of low-level vWF with ABO blood group system has also been documented in the literature. 32 About 80% of US residents with low vWF have “O” type blood group. 31 Furthermore, moderately low vWF levels and bleeding symptoms generally are not co-inherited within families and are not strongly associated with intragenic vWF mutations.32,33
Inherited PFDs constitute a large group of rare diseases involving a wide range of genetic defects that can lead to bleeding symptoms of varying severity. But relatively more common in communities where consanguineous marriages are more frequent like in the Middle East, India, and other developing countries.4,21,34,35 In our study, PFD is the fourth most common cause, that is 48 (12.8%), of bleeding manifestations. These include Glanzmann thrombasthenia (3.19%), defects of ADP receptor (3.0%), epinephrine receptor defect (3.0%), Bernard Soulier (1.0%), collagen receptor defect (0.8%), and unclassified (1.8%). We were not able to perform other tests such as platelet ATP secretion measured to rule out platelet release or storage pool defects.
Other rare inherited coagulation factor deficiencies have autosomal recessive inheritance in both genders. Hereditary FVII deficiency is the most common of the RICD.35,36 More frequent in countries like Iran, Middle East, and India, where consanguineous marriages are common. 36 Recently local study by Khalid et al also noted rare inherited bleeding disorders beside HA and HB. 37 Most of the patients with these deficiencies presented with mucosal bleeds like gum bleeding, epistaxis, easy bruising, and prolong bleeding after trauma; none of them had hemarthrosis. In this study, 3 patients presented with severe central nervous system (CNS) bleeding. These patients had severe FX and FVII deficiency and were treated with rFVIIa and FFP infusions. These RICD deficiencies account for 4.0% of all inherited bleeding disorders diagnosed in this study. This percentage figure is close to the figure found by Ahmed et al wherein they studied the prevalence of RICD in India. 4
Conclusion
Hemophilia A, vWD, and PFDs are common among congenital bleeding disorders. Availability of poor diagnostic facilities and lack of proper management for this group of patients, often lead to wrong diagnosis and inadequate treatment, as a consequence leading to crippling and at times life-threatening complications. Though development of inhibitors has not yet become a major issue but the rate of transfusion transmitted diseases particularly hepatitis C infection has gained a mammoth proportion. The acquisition of knowledge and expertise during this study will help in planning patient awareness program, establishment of hemophilia centers, and improvement in the management of patients with hemophilias across the country.
Footnotes
Acknowledgment
We would like to thank Novo Nordisk Hemophilia Foundation (NNHF) for their financial and technical support for Capacity Building of Hemophilia Care in Karachi region. Without their generous support, this study could not have been possible.
The author(s) declared no potential conflicts of interests with respect to the authorship and/or publication of this article.
Dr. Tahir Shamsi (Medical Director) has received grant from NovoNordisk Hemophilia Foundation.