
Abstract
Background
Moxonidine, an imidazoline I1 receptor agonist, is an effective antihypertensive drug that was shown to improve insulin sensitivity. RAAS-blockers are recommended as first-line therapy in patients with diabetes, alone or in combination with a calcium-channel antagonist or a diuretic.
Aims
This study compared the effects of moxonidine and ramipril on blood pressure (BP) and glucose metabolism in overweight patients with mild-to-moderate hypertension and impaired fasting glucose or type 2 diabetes.
Methods
Treatment-naïve patients for hypertension and dysglycemia were randomized to 12 weeks of double-blind moxonidine 0.4 mg or ramipril 5 mg once-daily treatment. At 12 weeks, for a further 12 weeks non-responders received combination of mox/ram, while responders continued blinded treatment.
Results
Moxonidine and ramipril were equivalent in lowering SiDBP and SiSBP at the end of the first 12 weeks. The responder rate was approximately 50% in both groups, with a mean SiDBP and SiSBP decrease of 10 and 15 mm Hg in the responders, respectively. The normalization rate (SiDBP < 85 mm Hg) was non significantly different between treatments groups. Moxonidine reduced heart rate (HR) (average −3.5 bpm, p = 0.017) during monotherapy, and when added to ramipril. HbA1c decreased significantly at Week 12 in both groups. Neither drug affected glucose or insulin response to the oral glucose tolerance test. In non-responders, moxonidine/ramipril combination further reduced BP without compromising metabolic parameters.
Conclusion
Moxonidine 0.4 mg and ramipril 5 mg were equally effective on BP lowering and were well tolerated and mostly metabolically neutral either as monotherapies or in combination. HR was lowered on moxonidine treatment.
Background
Risk factors such as diabetes, prediabetes and metabolic syndrome greatly exacerbate cardiovascular risk at any level of blood pressure (BP), and may attenuate the benefits that might otherwise be gained from lowering BP. The possibility that some antihypertensive drugs might be associated with the emergence of such risk factors is a matter of consideration in treatment choice.1–5 In particular, the long-term benefits of BP lowering may be compromised if antihypertensive drugs alter insulin sensitivity or promote the transition from pre-diabetes to overt diabetes. 2 Class differences in the metabolic effects of antihypertensives are therefore important to consider when choosing treatment for patients who exhibit metabolic disorders including prediabetes. Indeed, reducing the risk of incident diabetes is a major issue in people with prediabetes. 6
Use of angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) has been associated with a substantially lower risk of new-onset diabetes than placebo or active comparators. 7 Such an effect might contribute to the overall cardiovascular risk reduction achieved with drugs that act via the renin–angiotensin system. 8 Several clinical trials suggest that these drugs may have cardiovascular and metabolic advantages over drugs such as beta-blockers and diuretics 9 but this conclusion regarding cardiovascular events has not been proven definitively. However, the benefit of RAAS blockers is established in patients with diabetes and this class is clearly advocated as first-line therapy with a strong level of evidence. 10
Moxonidine is an effective antihypertensive drug which selectively targets imidazoline type-1 receptors and reduces central sympathetic nervous system activity.11,12 Moxonidine has been shown to improve insulin sensitivity in overweight patients with hypertension.13–15 High heart rate (HR) is a risk factor for cardiovascular events. 16 Interestingly, moxonidine, by reducing sympathetic activity and enhancing cardiovagal tone, has the potential to lower HR. However, such an effect on HR has not been clearly demonstrated in previous trials. In addition, cardiac autonomic dysfunction in patients with diabetes and prediabetes is mostly characterized by a vagal defect and a relative or absolute sympathetic activity enhancement.17,18 Two recent trials in hypertensive patients, INVEST and SPRINT, showed that high baseline and in-trial HR were strong predictors for cardiovascular outcomes, supporting the importance to consider HR in the management of patients with hypertension.19,20
Hypertensive patients with diabetes often require combination therapies to achieve tight BP control. 10 Second-generation central acting agents may be useful adjunctive therapy to achieve this goal. In most recent guidelines, treatment is recommended to be initiated with a combination of a RAAS blocker with a calcium channel blocker or thiazide-thiazide-like diuretic. 10 Importantly, combination of 2 drugs from different classes offers a greater BP reduction than doubling the dose of a unique drug. 21 If moxonidine is actually neutral or free of adverse metabolic effects it could be considered in combination with a RAAS blocker, specifically as an alternative to diuretics which may have deleterious effects on metabolism. There is no data on the combination of a RAAS blocker with a central acting agent such as moxonidine. Few studies have tested antihypertensive treatments in patients with prediabetes.22,23
The MARRIAGE (Moxonidine And Ramipril Regarding Insulin And Glucose Evaluation) study aimed to compare the effects of moxonidine and the ACE-inhibitor ramipril on hemodynamic and metabolic parameters in overweight patients with mild-to-moderate hypertension with impaired fasting glucose; or type 2 diabetes naïve for antihypertensive and glucose-lowering treatment. Recent guidelines have encouraged the use of combination anti-hypertensive treatment and emphasized the importance of HR in the hypertensive population. With this in mind, we decided to examine the effects of moxonidine and ramipril in the treatment of hypertension naive patients with prediabetes and diabetes.
Methods
Study Design and Patients
This was a prospective, randomized, double-blind, active-controlled, parallel-group, international, multicenter post-marketing surveillance study.
Patients aged from 18 to 79 years, with fasting plasma glucose (FPG) values ≥110 and <150 mg/dL, a body mass index (BMI) of >25.0 and <35.0 kg/m2, plus untreated known mild-to-moderate essential hypertension (standard office sitting systolic BP (SiSBP) ≥ 140 and <180 mm Hg and/or standard office SiDBP ≥85 and <110 mm Hg at Visit 1, confirmed at Visit 2) underwent a 2-week pre-screening placebo run-in period (Figure 1).

Study design. *The 2-week placebo run-in period was implemented only in patients with fasting plasma glucose values ≥110 and <150 mg/dL plus known mild-to-moderate hypertension.
Patients had impaired fasting glucose (defined as FPG levels ≥110 and <126 mg/dL [≥6.1 and <7.0 mmol/L]) or were considered as with diabetes if they had known diabetes (defined as FPG levels ≥126 mg/dL [≥7 mmol/L]6,24) treated with diet alone at Visit 1 and Visit 2. All patients provided written informed consent.
Patients were excluded from the study if they were requiring insulin or oral glucose-lowering agent, if their FPG levels were <110 mg/dL or > 150 mg/dL at Visit 1 and Visit 2 or if their glycated haemoglobin (HbA1c) were higher than 7.5% at Visit 2. Other exclusion criteria included severe hypertension (systolic blood pressure [SBP] ≥180 mm Hg or diastolic blood pressure [DBP] ≥110 mm Hg); unstable angina pectoris or recent (<3 months) myocardial infarction; a history of significant major organ-system disease, including but not limited to the cardiovascular, respiratory, gastrointestinal, renal, and hepatic systems and including neurologic and psychiatric diseases; and any known contraindication to the use of moxonidine. Pregnant and lactating women were also excluded from the study.
All included subjects proceeded to the active treatment phase. The planned duration of the active treatment phase was 24 weeks, comprising 12 weeks of randomized, double-blind single-agent treatment followed by assessment of response at Visit 4 (Figure 1). Patients who responded to treatment, defined as a reduction in sitting diastolic BP (SiDBP) of >10%, continued to receive the same single-agent antihypertensive treatment to which they had been initially randomized for a further 12 weeks in a double-blind manner, whereas non responders were switched to open-label combination therapy with both agents (Figure 1). Patients were followed for an additional 7–14 days after completing treatment which constituted a post-study follow-up phase. The study was conducted in accordance with the European and International guidelines for Good Clinical Practice (GCP). The CONSORT reporting guidelines wad used (Schulz KF, et al for the CONSORT Group. CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials). The study protocol, methods and documentation, including the informed consent form, were reviewed and approved by local Ethics Committees in the participating countries.
Treatment
Patients who satisfied the eligibility criteria were randomized (1:1) at Day 0 (Visit 2) to receive indistinguishable capsules of moxonidine 0.4 mg/day or ramipril 5 mg once daily for 12 weeks. At Visit 4, non-responders received a combination of moxonidine 0.4 mg and ramipril 5 mg once daily for the second and final 12-week period. Patients were instructed to take their medicine at the same time each morning (±1 h) and to omit their daily dose on the day of any study visit, to ensure that BP were measured at trough levels. Compliance with study medication was monitored by capsule count at each clinic visit. Patients were withdrawn from the study if compliance was <80% or >120% at two consecutive assessments.
Treatment with insulin or any oral glucose-lowering agents during the study was prohibited and had to be stopped at least 1 month before Visit 1. Treatment with antihypertensive agents other than study medication was also prohibited, and any patient requiring additional antihypertensive medication was to be withdrawn from the study. For patients receiving a statin, the dose of medication had to be kept constant during the study.
Primary Objective
The primary objective of the study was to demonstrate the equivalence of moxonidine and ramipril in reduction of SiDBP after 12-weeks double-blind therapy as compared to baseline. The equivalence margin was pre-defined in the protocol and set to 3 mm Hg.
Endpoints
The primary efficacy endpoint was the impact of moxonidine or ramipril on the change from baseline of SiDBP after 12 weeks. Secondary efficacy outcomes included the change from baseline to Week 24 in SiDBP, to Weeks 12 and 24 in SiSBP, as well as the proportions of patients responding to treatment (>10% reduction in SiDBP) or with BP normalization (ie, SiDBP <85 mm Hg) at Weeks 12 and 24. Changes from baseline to 12 or 24 weeks in plasma glucose and insulin levels at fasting and in response to the OGTT, in HbA1c and plasma lipids levels were also assessed. Safety endpoints included the frequency, severity, and seriousness of adverse events (AEs), as well as their causal relationship with study treatment.
Outcome Measurements
Arterial BP was measured at each study clinic visit by means of an automated device and applied principles of the Riva-Rocci method. All measurements for an individual patient at any given visit were to be made by the same staff member, on the same arm supported at heart level and using the same sized cuff. The patient was to sit quietly for at least 5 minutes before the first BP measurement. Three measurements were performed, at approximately 2-minute intervals, from which mean values were calculated. HR concomitant to the second BP measurement was recorded.
Blood samples were obtained at fasting at Visit 1 (pre-screening baseline), Visit 4 and Visit 6 to record metabolic efficacy parameters. Plasma glucose was measured at each visit according to standard methods implemented in local laboratories. At Visit 2, 4, and 6, the OGTT was performed with a solution containing 75 g of glucose. Other blood tests were performed at Visit 1, 4, and 6 and at the final study visit.
AEs were recorded from Visit 2 to the end of the study (Visit 7). Physical examinations and 12-lead electrocardiograms were also performed at pre-screening and the last study visit.
Statistical Analyses
Changes in SiDBP after 12 weeks of treatment were compared between the two treatments by means of analysis of variance (ANOVA) with treatment group and center as fixed factors. Equivalence of the two treatments would be concluded if the 95% confidence interval (CI) for the difference in mean change fell in the range −3 to +3 mm Hg. It was calculated that a sample size of 87 patients per group would be required to be secured of a statistical power of 80%. Assuming a drop-out rate of approximately 25% and 10% during the placebo run-in and active-treatment phases, respectively, it was determined that approximately 250 patients needed to be enrolled.
Because this was an equivalence study, the per-protocol (PP) population (defined as all patients with an assessment of BP at baseline and Week 12, and with no significant deviations from the protocol) was used for the primary efficacy analysis. The intent-to-treat (ITT) population of patients was defined as all patients with a BP assessment at baseline and at least one post-baseline BP measurement. The safety population (defined as all patients who received at least one dose of study treatment) was used for all safety analyses.
Within-group changes between visits in BP, HR, FPG and HbA1c levels were tested for significance by means of the one-sample t-test; mean changes in BP were compared between the moxonidine and the ramipril group by means of the two-sample t-test; response and normalization rates were compared between the two groups using Pearson's Chi-squared test; comparisons between non-responders and responders to either moxonidine or ramipril were done by means of ANOVA (BP, age, BMI, FPG and HbA1c levels) or Pearson's Chi-squared test (male/female ratio).
All analyses, tabulations, listings and graphs were done using the SAS® System version 8.2 (SAS Institute Inc., Cary, NC, USA) under Windows 2000 Professional.
Results
Patients’ Characteristics
Patients were recruited from 26 centers in France, Germany, Hungary and Spain between February 7, 2003 (first patient first visit) and May 27, 2004 (last patient last visit). In all, 243 patients were screened and 238 entered the placebo run-in period. Of these, 210 patients were randomized: 107 to moxonidine and 103 to ramipril (Figure 2). A total of 185 and 207 patients constituted the PP and ITT populations during the first 12-week treatment period. Baseline patient demographics and clinical characteristics were well balanced between treatment groups (Table 1).
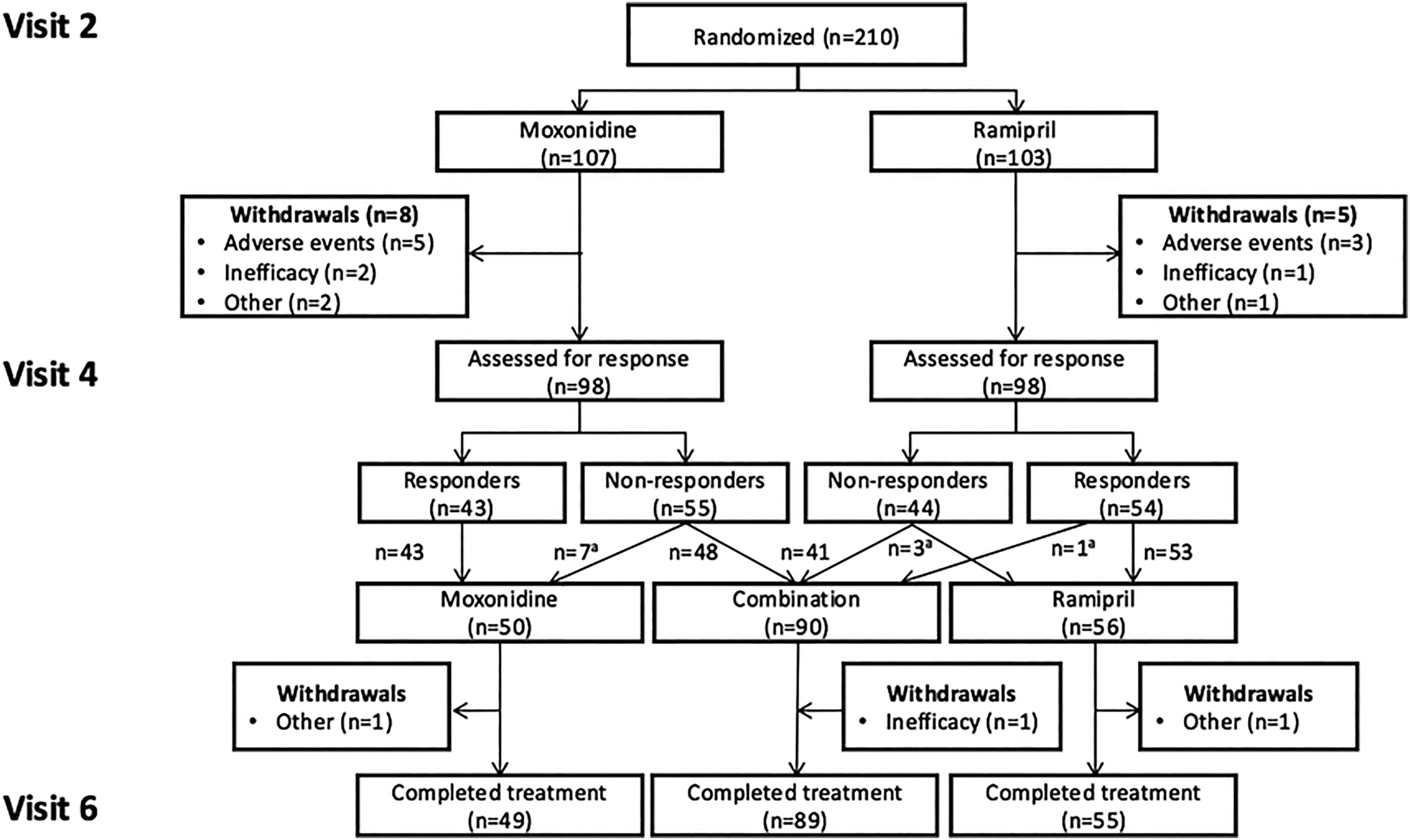
Patients disposition. aTreatment was misassigned in some patients.
Baseline Patient Demographics and Clinical Characteristics (Intent-to-Treat Population).

Data are given as numbers (%) or mean ± SD.
Abbreviations: BMI, body mass index; DBP, diastolic blood pressure; FPG, fasting plasma glucose; HbA1c, glycated hemoglobin; SBP, systolic blood pressure; SD, standard deviation.
Visit 2 values.
At baseline the distribution of comorbid conditions did not differ between treatment assignments, with the exception of spinal osteoarthritis (moxonidine, n = 11; ramipril n = 6). The distribution of concomitant medications was also similar between both groups, the most common being lipid-lowering agents (22.7% of patients).
Blood Pressure Response
During the First 12-Week Treatment Period
Trends in SiDBP (the primary efficacy endpoint) for the PP cohort (n = 185) are illustrated in Figure 3A, with additional data in Table 2A. The mean reduction from baseline in SiDBP exceeded 3 mm Hg with both treatments (p ≤ 0.001 vs baseline) (Figure 3A). The 95% CI for the difference between treatment groups fell within the prespecified range of −3 to +3 mmHg (Table 2A), thus meeting the condition for statistical equivalence between moxonidine 0.4 mg/day and ramipril 5 mg/day. Results in the ITT cohort (n = 207) were similar (Table 2A). SiSBP also decreased significantly in both treatment groups (−8.5 and −10.5 mean change from baseline, with moxonidine and ramipril, respectively) and there was no significant between-group difference in the PP cohort (<2 mm Hg between mean reductions) (Figure 3B and Table 2B). Similar data were observed in the ITT population.

Trends in blood pressure during the first 12-week treatment period in the per-protocol cohort (n = 185). Visit 1 was conducted at pre-screening (Week 2), Visit 2 was conducted at baseline (Week 0; randomization), Visit 3 was conducted at Week 6 and Visit 4 at Week 12. Data are shown as mean values with the standard deviation depicted by the vertical bars. (A) Trends in sitting diastolic blood pressure (SiDBP). Change from Visit 2 to Visit 4: Moxonidine * p < 0.001; Ramipril **p < 0.0005. (B) Trends in sitting systolic blood pressure (SiSBP). Change from Visit 2 to Visit 4: *** p < 0.0001 for both Moxonidine and Ramipril.
Changes in Blood Pressure During the First 12-Week Treatment Period.

Calculated on a censored PP cohort that excluded patients with SiDBP <85 mm Hg at baseline.
Responder and Non-Responder Patients
Responder rates for SiDBP in the PP cohort did not differ significantly in the 2 groups: 45.7% in the moxonidine group and 54.8% in the ramipril group, with similar data in the ITT cohort (Table 2A). The mean decrease in SiDBP and SiSBP in the responders’ groups was 10 and 15 mm Hg, respectively (Figure 4).

Change in sitting diastolic (SiDBP) and systolic (SiSBP) blood pressure, in the responder groups. Data as means ± SD. (A) SiDBP by visit and treatment in responder groups (ITT1 + ITT2). Change from Visit 2 to Visit 6: *p < 0.001, † p < 0.0001 (Mox-Mox: p = 0.0003; Ram-Ram p = 0.0001). Change from Visit 4 to Visit 6: ‡ p < 0.01 (Mox-Mox p = 0.0015; Ram-Ram p = 0.01). (B) SiSBP by visit and treatment in responder groups (ITT1 + ITT2). Change from Visit 2 to Visit 6: *p < 0.001, † p < 0.0001 (Mox-Mox p < 0.001; Ram-Ram p < 0.0001). Change from Visit 4 to Visit 6: The change in SiSBP was not significant in either treatment group (Mox-Mox p = 0.094; Ram-Ram p = 0.744).
Normalization rates for SiDBP (<85 mm Hg) were calculated on a censored PP cohort that excluded patients with SiDBP <85 mm Hg at baseline. The rates were above 40% in both treatment groups, with similar proportions for the censored ITT cohort (Table 2A).
BP in non-responder patients who were re-assigned to receive combination therapy was higher at Visit 4 (Week 12) than in those classified as responders to monotherapy who continued moxonidine and ramipril monotherapies. In addition, non-responder patients were slightly younger, and more males than responder patients who continued moxonidine or ramipril (Table 3).
Comparison of the Non-Responder and Responder Patients to Either Moxonidine or Ramipril Data at Visit 4 (Week 12).

Data are given as numbers (%) or mean ± SD.
Compliance rates during the second 12-week treatment period were never less than 95.8%.
During the Second 12-Week Treatment Period
After response assessment at Week 12, 196 patients continued in the study, of whom 50 continued moxonidine, 56 continued ramipril, and 90 were re-assigned to combination therapy. Unfortunately, 11 patients were misassigned, including 7 patients who were non responder to moxonidine and who were maintained on moxonidine monotherapy (Figure 2). A total of 193 patients constituted the ITT population during the second part of the study.
The changes in SiDBP and SiSBP during the 24-week treatment period for the responder groups are shown in Figure 4. The course of SiDBP (Figure 4A) was very similar in the two monotherapy groups. The maximum efficacy was attained after 3 months of treatment (mean 10 mm Hg decrease). Then SiDBP increased slightly during the following 3 months but still remained well below baseline values. These changes were statistically significant in both treatment groups (both p < 0.01). SiSBP (Figure 4B) decreased in a similar way in both responder groups at Visit 4 (mean 15 mm Hg decrease). Thereafter, SiSBP continued decreasing slightly in the ramipril responder group and increased slightly in the moxonidine responder group at Visit 5. Upon cessation of treatment, BP rose rapidly in both treatment groups during the 2-week follow-up period.
Combination therapy in patients classified as non-responders at Week 12 was associated with further reductions in SiDBP (Table 4). The mean SiSBP decrease during Weeks 12–24 was modest with no significant between-groups difference (Table 4). Normalization of SiDBP (<85 mm Hg) was observed in 68.6% of patients who completed 24 weeks of moxonidine monotherapy, 58.8% of patients who completed 24 weeks of ramipril monotherapy, and only 30.3% of patients who switched to combination therapy regardless of the initial monotherapy assignment.
Change of Sitting DBP and SBP in Responders and Non-Responders Groups Between Visit 2 and Visit 6 and Between Visit 4 and Visit 6 (ITT1 + ITT2 Samples).

Changes in Heart Rate
In the PP cohort, from baseline to Week 12 moxonidine was associated with a significant mean reduction in HR of 3.5 beats/min (p = 0.017) while in the ramipril group there was little alteration in HR (−0.6 bpm in means, p = 0.686). Between-groups difference was not significant (p = 0.139) (Table 5).
Changes in Heart Rate in the Per-Protocol Cohort (n = 185).

Abbreviations: HR, heart rate.
During the second 12-week treatment period, HR among patients maintained on monotherapy was generally stable. In patients switched from moxonidine to moxonidine-ramipril combination HR increased from 74.1 (±12.0) to 77.7 (±11.5) by an average of 3.7 beats/min (95%CI 0.3, 7.0; p = 0.0327) whereas HR decreased from 74.7 (±12.9) beats/min at Week 12 to 71.8 (±11.0) at Week 18 then 72.5 (±12.6) at Week 24 in patients switched from ramipril to ramipril–moxonidine combination. Between Week 12 and Week 24 the changes in HR between the two combination therapy groups differed significantly (5.7 bpm, p = 0.027) (Table 5).
Changes in Metabolic Parameters
Changes in Plasma Glucose and HbA1c
In the ITT population, FPG levels declined during the first 12 weeks period (Table 6), with no significant between-treatment differences. Plasma glucose response to the OGTT at Visit 2 and Visit 4 was similar in the two treatment groups (ITT cohort; Figure 5A), with no significant between-treatment differences.

Plasma glucose and insulin response during the oral glucose tolerance test (intention-to-treat population). Data as means ± SD. (A) Glucose response. Between Visit 2 and Visit 4, there was no relevant difference between the effects of the two study treatments on glucose AUC. The difference of 0.8% (p = 0.81) between treatment groups was not statistically significant. (B) Insulin response. Between Visit 2 and Visit 4, there was no relevant difference in treatment effect on insulin AUC. The difference of 3.3% (p = 0.69) between groups was not statistically significant.
Changes in Glucose and Insulin Levels.

Data are given as mean ± SD.
Abbreviations: AUC, area under the curve; FPG, fasting plasma glucose.
Plasma glucose response to the OGTT at Visit 6 was also similar across all treatment groups.
Mean HbA1c levels decreased significantly from baseline to Week 12 by 0.13% and 0.11% in the moxonidine (p = 0.0028) and the ramipril group (p = 0.0042), respectively. The difference in the changes between groups was not significant (Table 6). No significant changes in mean HbA1c were observed in any treatment assignment during the second 12-week treatment period.
Changes in Insulin Levels
At Visit 2 insulin response to OGTT was similar in moxonidine and ramipril groups. The changes in insulin levels during OGTT from baseline to Week 12 and from Week 12 to Week 24 were small and not significant across treatment arms (Table 6 and Figure 5B).
Changes in Lipid Levels
In both treatment arms there were small and non-significant increases in LDL-cholesterol, HDL-cholesterol and triglycerides from baseline to Week 24.
Safety
During the first 12-week treatment period, 5 (4.7%) moxonidine and 3 (2.9%) ramipril-treated patients withdrew from the study due to AEs. Treatment-emergent adverse events (TEAEs) were reported in 31 patients (29.0%) randomized to moxonidine and 19 patients (18.4%) randomized to ramipril. Between-group differences were mostly attributable to a greater incidence of dry mouth in the moxonidine group (in 13 patients vs 1 patient in the ramipril group). Eight TEAEs were linked with ramipril use, of which the most common were abdominal pain and headache. One serious AE was reported in each treatment group, neither was considered to be associated with use of study medication (Supplementary table).
During the second part of the study (Weeks 12-24), there were no premature discontinuations due to AEs. TEAEs were reported in 24 patients (12.2%), of which 5 of them could be treatment attributable. The only serious AE was not considered treatment related (supplementary table).
Discussion
The RAAS blockers are a reference treatment in hypertensive patients with diabetes and prediabetes. In this study we compared two antihypertensive drugs: moxonidine, a centrally acting imidazoline I1 receptor agonist 11 ; and ramipril, an ACE inhibitor in overweight patients with mild to moderate hypertension and prediabetes or type 2 diabetes. These two drugs have proved to be equally effective in lowering BP (SiSDB and SiSBP). They were a high proportion of responders, and the mean decrease in responders was 10 mm Hg for SiDBP and 15 mm Hg for SiSBP. HR was lowered on moxonidine treatment. No noticeable changes were observed in metabolic parameters, including the response to the OGTT, except for FPG and HbA1c which decreased mildly but significantly and similarly in the two groups after 12 weeks of monotherapy.
Blood Pressure Effects
Both moxonidine and ramipril were found to be statistically equivalent in their ability to lower SiDBP over 12 weeks at the doses investigated, and SiSBP decrease was also similar. The unexpected BP increase with monotherapies during the second 12-week period could be explained by the number of non-responders that were misassigned to monotherapy (7/50 in moxonidine group, 3/54 in ramipril group) at Visit 4. Overall, BP reduction on monotherapy by moxonidine or ramipril was consistent with a normalization rate for SiDBP (<85 mm Hg) of around 45% for both during the first period and a further improvement during the second one (68.6% and 58.8%, respectively). However, in both monotherapy groups, mean SiSBP in responders remained upper than the goal of 130 mm Hg or lower recommended in the recent guidelines. 10 The 7 non-responders to moxonidine who were misassigned to continue on moxonidine monotherapy might have reduced the benefit expected during the second period. Mostly, the non-achievement of BP target might be because nearly half of the patients actually had diabetes according to current diagnostic values (FPG ≥126 mg/dL (7.0 mmol/L)), 24 and hypertension is more difficult to treat in patients with diabetes, mostly requiring combination of two to three agents to reach target. 25
The additional BP reduction with combination therapy during the second 12-week treatment period was modest compared with the effects observed with combination of moxonidine and amlodipine. 26 The addition of one drug is supposed to induce a further decrease in SiDBP by an amount roughly similar as their respective effect when taken solely during the first period. In fact, the SiSBP decrease was markedly lower in the group who received combination therapy. However, the patients who received combination therapy were confirmed non-responders to moxonidine or ramipril, and studies have shown that non-responders to any antihypertensive drug will require at least one more drug. 27 To note, the difference in baseline characteristics of this non-responders subgroup compared with the responders (higher baseline BP and younger) suggests a difficult-to-control patient profile for which combination of at least two drugs with at least one RAAS blocker might be necessary to achieve the recommended goal. 10
The HOT study 28 aimed to assess the optimum diastolic BP in patients with hypertension and a DBP between 100 and 115 mm Hg. Larger BP decrease than in our study is probably due to the higher BP levels at baseline. HOT study showed that intensive BP lowering was associated with a reduction in the rate of major cardiovascular events and cardiovascular mortality. The maximum benefits were observed for on-treatment SBP between 130 and 140 mm Hg, and DBP values between 80 and 85 mm Hg. In particular, in the patients with diabetes there was a significant 51% reduction in the composite cardiovascular outcome (nonfatal myocardial infarctions, nonfatal strokes, and cardiovascular deaths) in target group with DBP ≤ 80 mm Hg compared with target group with DBP ≤ 90 mm Hg, which supports using multiple combination therapy if needed in these patients. We here show that such a combination can be effective for lowering BP in patients with diabetes or prediabetes without major metabolic effects and serious AEs. In addition, when initiating on combination therapy, BP goal was shown to be achieved earlier and reduction of CV outcomes to be stronger. 29
Heart Rate Changes
HR was lower on moxonidine treatment compared with ramipril in monotherapy or added to ramipril. HR decrease on moxonidine was likely to result from autonomic activity modulation. The sympathetically-induced tachycardic responses in spontaneously hypertensive rats was shown to be inhibited by moxonidine, 30 and it has been suggested that moxonidine may have BP-independent cardioprotective effects, such as a reduction of the risk of atrial fibrillation recurrence after ablation treatment. 31 Another study showed in rats that high moxonidine doses were strongly parasympathomimetic through the activation of central alpha2-adrenoceptors. 32 In a double-blind, placebo-controlled study in untreated hypertensive patients, after a single 0.4-mg moxonidine dose HR decreased during the night hours. 33 In healthy individuals a single moxonidine dose increased cardiovagal tone and decreased cardiac sympathetic activity. 34 A 6-month clinical trial showed that the effect of moxonidine in reducing insulin resistance in patients with metabolic syndrome depended on the severity of hypersympathicotonia manifesting in HR >80 bpm at rest. 35
It is now clear that lowering a high HR reduces significantly the risk of CV outcomes.16,19,20 In the INVEST study which included hypertensive patients with coronary artery disease, HR predicted adverse outcomes, and on-treatment HR was more predictive than baseline HR. 19 In the SPRINT study, 20 a higher baseline resting HR was associated with a higher cardiovascular risk, this increased risk being greater in subjects on intensive than on standard treatment. These observations are in accordance with the ESC-EASD recommendation to consider HR >80 bpm as a CV risk 10 and suggest that HR reduction using antihypertensive drugs may be beneficial in subjects with hypertension. 20 The LIFE study also illustrates that persistence or development of HR >84 bpm was associated with an 89% greater risk of CV death and a 97% increase of all-cause mortality. 36 In this context, the lowering effect of moxonidine on HR is interesting to consider. Evidence supports the use of RAAS blockers in patients with diabetes,10,37 and BP control often requires multiple therapy with a RAAS blocker and a calcium channel blocker or a diuretic. 10 Sympathetic activity was clearly shown to be enhanced in patients with diabetes 17 and in obese patients with hypertension. 18 Thus, central sympatholytic drugs are logical in these patients and embody a potential interest for hypertensive patients with elevated HR.
Changes in Metabolic Parameters
In this population with prediabetes or well-controlled diabetes, there was a modest but significant HbA1c reduction without clinically significant change in lipids on moxonidine or ramipril treatment, in monotherapy nor on combination therapy. Glucose and insulin responses to the OGTT did not change significantly and were similar after 12 or 24 weeks across the treatment groups. These results are in line with previous studies showing neutral or beneficial effects of ACE-inhibitors and moxonidine on glucose profile and insulin sensitivity. Moxonidine has been associated with improvements of insulin sensitivity in overweight patients with hypertension.13–15 In prediabetes, the DREAM study also showed that ramipril significantly increased regression to normoglycemia. 38
Strengths and Limitations
This study has some strengths. First, this is a randomized controlled study including an active comparator, a RAAS blocker, the ACE-inhibitor ramipril, as recommended as first-line therapy in the current guidelines. Second, the study population was homogeneous since only patients with type 2 diabetes or prediabetes, all naïve for antihypertensive and glucose-lowering treatment, were included. Third, glucose metabolism was assessed by glucose and insulin measurements both at fasting and after an OGTT. Fourth, we measured HR which has never been studied extensively during trials.
However, the study also has some limitations. First, we included patients with well-controlled diabetes or prediabetes, and thus the conclusion may not be extended to patients with poorly-controlled diabetes. Moreover, subjects enrolled in this study were overweight or obese (with a BMI 25-34.9), like in the previous studies which tested moxonidine.13–15,33,35,39 Thus, the conclusion may not be extended to hypertensive subjects with normal body weight or with severe or massive obesity. Second, this study was conducted at the same time as the HOT study, approximately 20 years ago, 28 with subsequent changes in diagnostic criteria for diabetes which now require two FPG measurements. In addition, SBP is now the primary target for management of hypertension. However, the effects of different approaches on DBP should not be ignored. The principal goal at that time was to lower the SiDBP under 85 mm Hg or have a reduction of the SiDBP by >10%. The SiSBP was assessed but wasn’t the principal goal while in 2019 ESC-EASD guidelines targets are now a SBP between 120 and 130 mm Hg, and a DBP between 70 and 80 mm Hg.10,37 Third, HR was measured only once at each visit using automated BP devices. The effects of moxonidine on HR need to be confirmed in further trials and assessed using electrocardiography and better 24-hour recordings. Finally, the statistical plan defined at the initiation of the study presents some limitations. At the time of writing the protocol, we decided to perform a standard analysis for comparison of endpoints such as BP between the moxonidine and the ramipril group by using a two-sample t-test because it was the easiest way to understand the statistical analysis, and so, this is the analysis presented in this paper. Changing the statistical analysis after the study database had been unblinded would have been a violation of ICH guideline E9 Statistical Principles for Clinical Trial and so was not performed, but possibly more informative analyses like a two-way repeated measures ANOVA might have been considered.
Conclusion
Moxonidine 0.4 mg and ramipril 5 mg once daily are safe, well tolerated and equivalently effective antihypertensive treatments in overweight patients with mild-to-moderate hypertension and impaired fasting glycemia or diabetes. In case of insufficiently effective monotherapy, both agents can be combined to achieve further BP control without compromising metabolic parameters. The slight lowering HR effect of moxonidine is interesting to consider due to the worse CV outcomes related to high HR, but due to the limitation of the study's design, this result needs to be confirmed with other studies. Moxonidine may be considered as a useful adjunctive therapy for hypertension in dysglycemic patients.
Supplemental Material
sj-docx-1-cpt-10.1177_10742484241258381 - Supplemental material for MARRIAGE: A Randomized Trial of Moxonidine Versus Ramipril or in Combination With Ramipril in Overweight Patients With Hypertension and Impaired Fasting Glucose or Diabetes Mellitus. Impact on Blood Pressure, Heart Rate and Metabolic Parameters
Supplemental material, sj-docx-1-cpt-10.1177_10742484241258381 for MARRIAGE: A Randomized Trial of Moxonidine Versus Ramipril or in Combination With Ramipril in Overweight Patients With Hypertension and Impaired Fasting Glucose or Diabetes Mellitus. Impact on Blood Pressure, Heart Rate and Metabolic Parameters by Paul Valensi, MD and Selim Jambart, MD in Journal of Cardiovascular Pharmacology and Therapeutics
Footnotes
List of abbreviations
Acknowledgements
This article was written with the support of Abbott laboratory. The authors would like to thank all of the members of the scientific committee for the design of the study and the ethics committees for the supervision of the study. They thank the 26 participating centers and the 31 investigators in Germany, Hungary, Spain, and France without whom this study would not have been possible. And they also thank KPL and Galien Health Publishing for the medical writing assistance for the preparation of this manuscript.
Authors’ Contributions
Authors PV and SJ designed the study, were fully involved in the study and contributed to the methodology. PV wrote the manuscript. SJ provided resources and analysis. PV and SJ reviewed the manuscript and contributed to the discussion. PV is the study guarantor with full access to all the data in the study and accept responsibility for the integrity of the data and the accuracy of the data analysis. All authors read and approved the final manuscript.
Availability of Data and Materials
The datasets generated during and/or analyzed during the current study, under the General Data Protection Regulations (GDPR) may be available for research collaboration purposes upon reasonable request to the corresponding author (Paul Valensi, paul.valensi@noos.fr) and will require the completion of a data processing agreement.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: PV discloses the following potential conflicts of interest: lectures for Abbott, AstraZeneca, Bayer, Eli Lilly, Hikma Pharmaceuticals, Merck Sharp & Dohme, Novo Nordisk, Novartis, Pfizer, Sanofi, Servier; research grants from Abbott, Bristol-Myers Squibb–AstraZeneca, Novo Nordisk; participation in expert committees for AstraZeneca, Boehringer Ingelheim, Novo Nordisk, Daiichi Sankyo, Sanofi, Servier.
Ethics Approval and Consent to Participate
The study protocol, amendment, elucidation, the written subject informed consent form, subject procedures and any other written information provided to the subject were submitted to the Ethics Committee (EC) at each study center. Written approval was obtained for all centers before the commencement of the study.
The study was conducted in accordance with the European directive 91/507/EEC and the International Conference on Harmonization (ICH) consolidated guidelines for Good Clinical Practice (GCP) dated July 17, 1996, which originated from the ethical principles laid down in the Declaration of Helsinki. Solvay Pharmaceuticals policies and procedures were followed.
In Germany, the study was approved by the Independent Ethics Committees: Ethics Committee of the “Charite Berlin Campus Berlin-Buch” (Berlin), Ethics Committee of the “Landesärztekammer Hessen”, Ethics Committee of the “Landesärztekammer Baden-Württemberg” (Stuttgart), Ethics Committee of the “Landesärztekammer Rheinland-Pfalz” (Mainz) and Ethics Committee of the “Ärztekammer Berlin” (Berlin).
In Hungary, the study was approved by the Central Ethics Committee – Medical Research Council Ethics Committee for Clinical Pharmacology (Budapest) and by the following Local Ethics Committees: Regional Committee of Science and Research Ethics (Miskolc), Regional Committee of Science and Research Ethics (Györ), – Institutional Committee of Research Ethics (Budapest), Institutional Ethics Committee (Budapest) and the Ethical Committee of Pharmamedcor Surgeries (Budapest).
In Spain, the study was approved by the Independent Ethics Committees: Clinical Research Ethics Committee of the Hospital Clinico Universitario San Carlos from Madrid (Madrid), Clinical Research Ethics Committee of the Hospital “Virgen Macarena” from Sevilla (Sevilla), Clinical Research Ethics Committee of the Hospital Clinico Universitario “San Cecilio” from Granada (Granada), Clinical Research Ethics Committee of the Autonomous Community of Andalucia (Sevilla), Clinical Research Ethics Committee of Galicia (Santiago de Compostela) and Clinical Trials Agency of the Hospital Clinic I Provincial from Barcelona (Barcelona).
In France, all study centers were approved by the Independent Ethics Committee: Comité Consultatif de Protection des Personnes dans la Recherche Biomédicale, Hôpital Robert BALLANGER, Centre Daniel Eisenmann (Consultative Committee for Protection of Persons who Participate in Clinical Research).
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by Solvay pharmaceuticals and Abbott.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.