
Abstract
Cardiovascular disease (CVD) is the leading cause of death in the United States and worldwide. A major risk factor for this condition is increased serum low-density lipoprotein cholesterol (LDL-C) levels for which statins have been successful in reducing serum LDL-C to healthy concentrations. However, patients who are statin intolerant or those who do not achieve their treatment goals while on high-intensity statin therapy, such as those with familial hypercholesterolemia, remain at risk. With the discovery of PCSK9 inhibitors, the ability to provide more aggressive treatment for patients with homozygous and heterozygous familial hypercholesterolemia has increased. Ezetimibe reduces LDL-C by 15%-20% when combined with statin. 2,3 Protein convertase subtilisin/kexin type 9 (PCSK9) inhibitors have been found to achieve profound reductions in LDL-C (54%-74%) when added to statins. They have shown dramatic effects at lowering major adverse cardiovascular events (MACE) in high-risk patients 4 with LDL-C levels ≥70 mg/dL and can be used in populations that are statin intolerant or not at goal levels with maximally tolerated statin therapy. PCSK9 inhibitors also produce minimal side effects. Myopathy, a common side effect for patients on statins, has been rare in patients on PCSK9 inhibitors. Randomized trials have shown that reduction in LDL-C has translated to clinical benefits even in patients who have not achieved their LDL-C target.
Keywords
Introduction
Despite the availability of therapies that reduce low-density lipoprotein cholesterol (LDL-C), cardiovascular disease still remains the leading cause of mortality and morbidity. 1 Proprotein convertase kexin/subtilisin type 9 (PCSK9) contributes to these cardiovascular events by increasing plasma levels of LDL-C through its disruption of the LDL receptor’s (LDLR) endocytic recycling pathway and the subsequent lysosomal degradation of this receptor. This inhibits the uptake of LDL-C, leading to hypercholesterolemia. 2 PCSK9 inhibitors reduce cardiovascular risk 3 by preventing this cycle. 4
In 2003, “gain of function” mutations on the newly discovered PCSK9 gene were associated with autosomal dominant hypercholesterolemia, a causative risk factor for cardiovascular disease. 5 A study in 2005 further corroborated the relationship between hypercholesterolemia and PCSK9 by relating low LDL-C levels in a group of African Americans with “loss of function” nonsense mutations on the PCSK9 gene. 6 These studies opened the floodgates on PCSK9 research with subsequent research devoted to better understanding of PCSK9 and its use in treating cardiovascular disease.
Structure of PCSK9
PCSK9 is a serine protease composed of 692 residues. It contains a prodomain, catalytic domain, and a histidine rich C-terminal domain; the PCSK9 zymogen undergoes autocatalytic cleavage between the pro and catalytic domain before secretion into the extracellular matrix. However, the non-covalently bound prodomain shepherds the catalytic domain into the plasma, and stays adhered to it throughout its lifetime, blocking the enzymatic pocket. PCSK9’s proteolytic activity is unnecessary for its effects on LDL-C. 7,8
Once secreted, the PCSK9 complex binds to the first epidermal growth factor-like repeat (EGF-A) of the LDLR’s EFG domain. 8 A crystal structure of the PCSK9-LDLR ectodomain showed that the primary interactions occur over a 1000 Å2 hydrophobic patch with polar interactions along the periphery (Figure 1). 9,10 Binding is calcium dependent and increases with reduced pH. 8 In fact, the low pH of the lysosome increases the strength and number of interactions between LDLR and PCSK9; PCSK9 has a 150-fold increased infinity for LDLR at pH 5.3 compared to pH 7.4. 11 Studies have also shown that autocleaved PCSK9 can bind to LDLR intracellularly. The result of this interaction is the degradation of both proteins. This leads to lower LDLR levels and subsequently increasing concentrations of circulatory LDL.
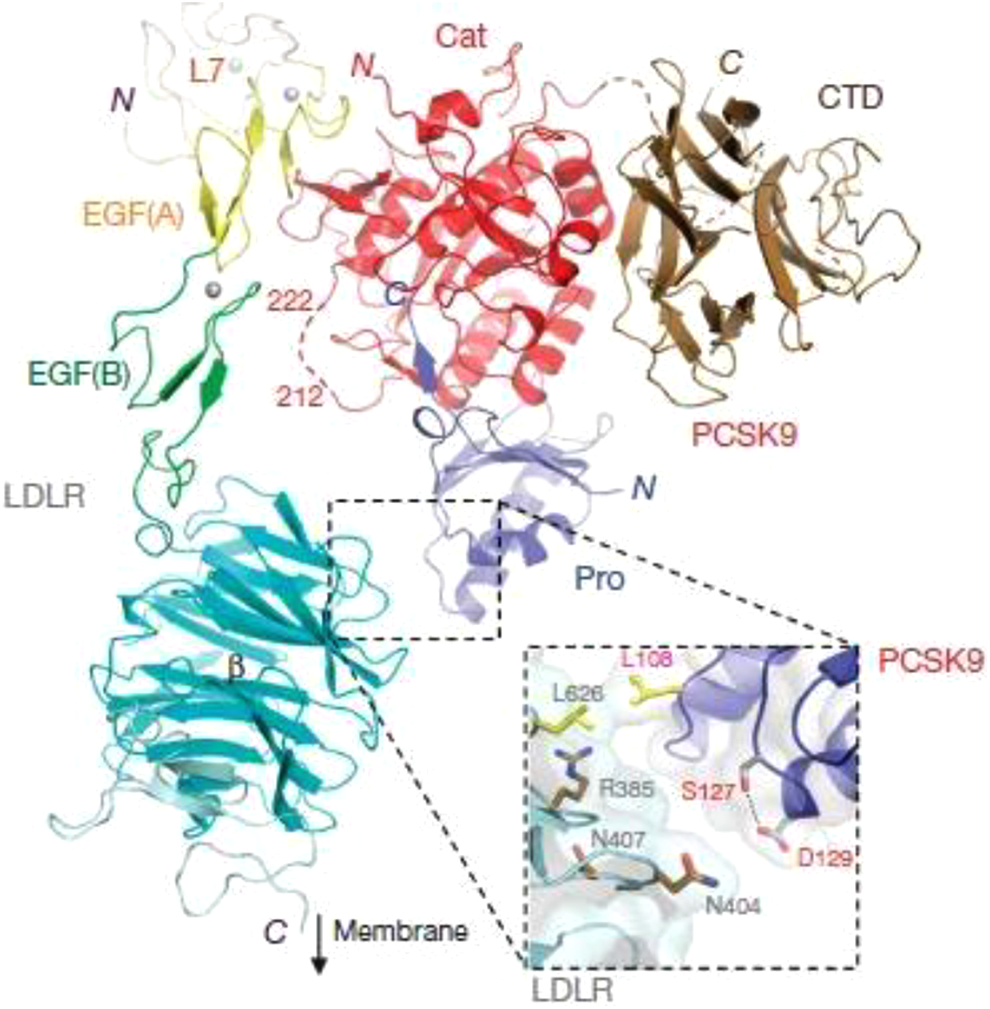
Crystal structure of PCSK9-LDLR structure. 9
In addition, 30% to 40% of plasma PCSK9 molecules are bound to LDL at any given point in time. In vitro studies of LDL-PCSK9 interaction patterns have shown that several gain of function mutations associated with familial hypercholesterolemia (FH) inhibit LDL-PCSK9 binding, leading to the conclusion that LDL-PCSK9 binding acts as a form of PCSK9 inhibition. A disordered N-terminus region in PCSK9’s prodomain was determined to be a necessary component for this interaction with the presumed binding site being an amphipathic alpha helix in the region. 12
PCSK9 and LDLR Interaction Patterns
PCSK9 and LDLR interactions can be classified into several subcategories. The first is parallel expression: Sterol Regulatory Element Binding Proteins (SREBP) activation causes the transcription of PCSK9 and LDLR. Therefore, parallel expression describes the physiological activity of SREBP in which an increase of PCSK9 occurs concurrently with an increase of LDL receptors. Conversely, parallel degradation describes a simultaneous decrease of PCSK9 and LDLR levels. Parallel degradation normally occurs when PCSK9 interacts with LDLR, which causes the complex to be engulfed and thereby degraded via lysosomes. Parallel expression and parallel degradation are widely understood and studied interactions between PCSK9 and LDLR receptors. Low levels of PCSK9 can have several causes, including therapies causing PCSK9 inhibition and natural loss-of-function mutations. Low levels of PCSK9 cause high concentrations of surface LDLR since there is less PCSK9 available to drive parallel degradation. This decreases PCSK9 levels even further since the molecules that do bind to LDLR are eventually degraded. Thus, such a situation described has come to be known as high LDLR reciprocal regulation. 13
Mode of Action of PCSK9 Inhibitor
A variety of PCSK9 inhibitors have been developed to inhibit PCSK9 at different stages in its life cycle. Inhibition mechanisms can be classified under 3 different groups: (1) LDLR binding inhibition, (2) PCSK9 synthesis inhibition, and (3) inhibition of auto-catalytic processing. 7 LDLR binding inhibition prevents PCSK9 from binding to the LDLR, allowing for a greater number of receptors to be recycled to the cell surface for further LDL-C removal. PCSK9 synthesis inhibition involves preventing PCSK9 formation at the level of translation, silencing the PCSK9 gene. The inhibition of autocatalytic processing involves interrupting the autoprocessing of PCSK9 thereby prohibiting maturation and cell secretion. 14
The PCSK9 Mediated Degradation of LDLR Pathway shows PCSK9 binding to the extracellular region of the LDLR. The entire complex is then taken into the cell. PCSK9 prevents recycling of the LDLR and the complex is transferred to an endosome where the entire complex is degraded. The LDLR Regeneration Pathway takes place with no interference from PCSK9, resulting in the degradation of LDL-C. The apolipoprotein B100 (ApoB100) portion of LDL-C binds to the ligand-binding domain of LDLR. After endocytosis, the LDLR/LDL-C complex undergoes acid-dependent release. The LDLR is then recycled back to the cell surface, allowing for further uptake of LDL-C (Figure 2). 12

LDLR regeneration and PCSK9 mediated degradation of LDLR pathways.
Non-LDL-C-Lowering Effects of PCSK9 Inhibition
In addition to increasing LDLR concentrations and lowering LDL-C concentrations, some PCSK9 inhibitors have been found to decrease apolipoprotein B (apoB) and very-low-density lipoprotein (VLDL) cholesterol. Since not all PCSK9 inhibitors decrease VLDL, there may be differing effects that lie outside the shared commonality of LDL-C reduction. PCSK9 inhibitors also minorly increase high-density lipoprotein cholesterol (HDL-C) and apolipoprotein A-I (apoA-I). One explanation for this effect is that reduced apoB lowers the amount of cholesterol that is transferred from HDL to particles containing apoB. 15
PCSK9 inhibitors of varying classes have also been found to reduce lipoprotein(a) (Lp(a)) levels. Statins, by contrast, have no effect on Lp(a). Lp(a) is a causal risk factor for cardiovascular disease and more than 20% of the world has elevated circulating Lp(a) levels. 15 There are multiple hypotheses about how PCSK9 inhibitors reduce Lp(a). One hypothesis suggests that Lp(a) competes poorly with LDL for LDLR binding. Thus, when PCSK9 inhibitors increase LDLR expression and particularly when LDL levels are low, Lp(a) decreases. 16 Other explanations include decreased apoB or Lp(a) synthesis. 17
Therapeutic PCSK9 inhibition has not been associated with an increase in diabetes. 18 Inhibition occurs primarily inside the liver, without affecting other parts of the body where PCSK9 is produced. However, PCSK9 loss of function mutations have been shown to cause a modest increase in the onset of diabetes. 19 Genetic PCSK9 deficiency increases plasma glucose levels and therefore the risk for type 2 diabetes. A study using PCSK9 knockout (KO) mice found that complete absence of PCSK9 caused increased glucose intolerance and impaired insulin secretion, while having no effect on insulin resistance in pancreatic β-cells. 20 This, combined with the fact that type 2 diabetes is less common in subjects with familial hypercholesterolemia, lends support to the idea that LDLR plays a role in the development of diabetes. 21
Finally, reduced PCSK9 levels have been associated with lower levels of thrombosis. In PCSK9 KO mice, a clear reduction in FeCl3 injury-induced carotid artery thrombosis was determined. 22 Meanwhile, higher PCSK9 levels are associated with higher platelet reactivity in patients with acute coronary syndrome (ACS). In the PCSK9-REACT Study, PCSK9 levels and platelet reactivity were analyzed from ACS patients on daily doses of an antiplatelet medication, either prasugrel or ticagrelor. They found a direct correlation between circulating PCSK9 levels and platelet reactivity for every participant, which translated into more atherothrombotic events at the 1-year mark for those with higher serum PCSK9. 23 These findings show that PCSK9 inhibition does more than simply lower LDL-C, and there may be multiple factors that contribute to its ability to prevent and treat cardiovascular disease (CVD).
Role of PCSK9 Inhibitors in Familial Hypercholesterolemia
Familial hypercholesterolemia is a genetic disorder which results in extremely elevated plasma LDL-C levels and increased risk of early onset CVD. FH can be either heterozygous (HeFH) or homozygous (HoFH), depending on if there is one mutated allele or two, respectively. Mutations that cause FH tend to occur on the gene which encodes LDLR but can also occur on PCSK9, apoB, or LDLR adaptor protein genes. 24 Because of this, many treatments that rely on LDLR function have reduced efficacy in FH patients. 25 It is important to note that some HeFH patients have additional mutations in other alleles that cause hypercholesterolemia. This is called compound heterozygosity and can lead to much higher LDL levels, similar to those of homozygous patients.
HoFH patients often have serum LDL-C levels of >500 mg/dL. 26 Concurrent regimens of multiple drugs are the most effective means of lowering cholesterol in these patients. An 80 mg/day dose of atorvastatin has been shown to lower LDL-C levels by 28% in subjects with homozygous FH via a reduction in overall cholesterol synthesis. Even LDLR receptor negative patients experienced the same reduction. 27 Ezetimibe in combination with statins can reduce LDL-C levels by an additional 10%-15%. 28,29 HoFH is also independently associated with elevated PCSK9 levels; in the TESLA study, the addition of the PCSK9 inhibitor evolocumab to a stable lipid-reducing therapy decreased LDL-C by 30% compared to placebo at the 12-week mark. 30 Similarly, the ODYSSEY HoFH trial reported a 36% LDL-C reduction after 12 weeks of treatment with alirocumab. 31 Both studies also noted significant reductions in other atherogenic lipids including apoB and Lp(a). Although these reductions are not as robust as in people without HoFH, they represent the first line of treatment for the disease.
Monoclonal Antibodies
The therapeutic agents furthest in development are monoclonal antibodies (mAbs). MAbs are monospecific antibodies derived from clones of a single parent cell. These antibodies can be further classified as either murine, chimeric, humanized, or fully human, depending on the percentage of human genetic material used. MAbs can be taken in conjunction with statins, resulting in increased LDL-C reduction. 32,33 In July 2015, evolocumab (AMG-145) was the first PCSK9 monoclonal antibody to get EMA (European Medicines Agency) approval for the treatment of primary hypercholesterolemia. 34 It was approved by the FDA as a treatment option for those who cannot meet target LDL-C levels shortly after. 35 Evolocumab is a fully human antibody that inhibits PCSK9 by binding to the relatively flat region of PCSK9, blocking PCSK9 interactions with LDLR. 36 In the OSLER-1 and OSLER-2 trials, evolocumab’s efficacy in lowering LDL-C was studied in 4465 patients who had already taken part in phase II and III trials. The OSLER trials’ participants underwent a year of monthly or biweekly doses of evolocumab supplemented by standard therapy, or standard therapy alone. In both trials the drug was administered subcutaneously. 37 The Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk (FOURIER) trial studied 27,525 patients. They found the use of evolocumab in conjunction with statin therapy decreased and sustained LDL-C levels by 59% after 48 weeks, compared to placebo. Injection site reactions were the only adverse event to be significantly higher in the evolocumab group versus the placebo group (2.1% vs. 1.6%, P < .001, respectively), and these reactions were mild. 18
Alirocumab was approved by the FDA as a mAb PCSK9 inhibitor within a week of evolocumab’s European approval. 38 The ODYSSEY LONG TERM trial administered biweekly injections of alirocumab (150 mg) or placebo to patients on maximally tolerated statins. After 78 weeks it found alirocumab lowered LDL-C levels by 62% versus placebo. Adverse events were comparable to trials with evolocumab. 39 The ODYSSEY OUTCOMES trial evaluated the effect of alirocumab on patients who had ACS in the 1 to 12 months prior to enrollment with similar results. LDL-C levels were reduced by 62.7% compared to placebo after 4 months and remained 54.7% lower than the placebo group after 48 months. Injection site reactions with alirocumab were higher than in the placebo group (3.8% vs. 2.1%, respectively). 40
Although their safety profiles are comparable, a meta-analysis found evolocumab to be a more potent reducer of LDL-C than alirocumab. Biweekly or monthly doses of evolocumab were found to be 14%-20% more effective at reducing LDL-C than weekly, biweekly, or monthly doses of alirocumab. The study also concluded that the addition of PCSK9 inhibitors to statin therapy reduces LDL-C by 54%-74% compared to placebos and by 26%-46% compared to statin therapy plus ezetimibe. 41 Although often used in conjunction with statins, it is important to note that monoclonal antibody PCSK9 inhibitors are also effective as stand-alone treatments. The DESCARTES trial demonstrated evolocumab reduces LDL-C levels by 55.7% ± 4.2% compared to placebo for patients on a background therapy of diet alone. 42 This reduction is significant for those intolerant to statins or unable to reach LDL-C goals with statins or ezetimibe.
Bococizumab is a humanized mAb that underwent phase 3 trials but has since been discontinued. It was initially very efficient at lowering LDL-C but was found to produce antidrug antibodies in 48% of subjects at the 1-year mark in a multinational study. Moreover, high levels of antidrug antibodies were associated with significant reductions in LDL-C-lowering effects. 43 A new humanized mAb, LY3015014, has not had these problems and is longer lasting than evolocumab and alirocumab. In a phase 2 study, subcutaneous administration of LY3015014 produced LDL-C reductions of up to 50.5% compared to placebo with very minor adverse events, the most common being injection site reactions. Anti-drug antibodies were present with slight differences in the LY3015014 and placebo groups (6.5% vs. 4.7%) 44 but remained substantially lower than trials with bococizumab. 1B20 is another new mAb currently under investigation. It binds to PCSK9 with sub-nanomolar affinity and acts as an in-vitro antagonist, disrupting PCSK9-LDLR protein interactions and completely blocking PCSK9 inhibition on cellular LDL uptake. It lowers plasma LDL-C levels by 50%-70% in CETPtg(LDLR+/-) mice and can also be used in conjunction with statins. In metabolic syndrome monkeys, the addition of simvastatin to 1B20 resulted in a 40% decrease in plasma LDL-C levels. 1B20 alone caused a 20% decrease in LDL cholesterol. 45
siRNA
Small interfering RNA (siRNA) technology is an upcoming and relatively new tool which has also been adapted to PCSK9 inhibition. SiRNA are double-stranded, non-coding RNA which are composed of a passenger strand and a guide strand. They silence genes by selectively impeding the translation of messenger RNA (mRNA) complementary to the guide strand. The chemically synthesized siRNA inclisiran is at the forefront of PCSK9 silencing siRNA developments and has successfully undergone phase 3 trials. It is currently approved for use in the EU and US. 46,47 Inclisiran binds to the mRNA that code for PCSK9 with the help of an RNA-inducing silencing complex (RISC). Once paired, RISC activates RNase and cleaves the mRNA, preventing PCSK9 protein formation (Figure 3). This mechanism remains active even after the mRNA has been degraded and is one explanation for inclisiran’s long efficacy. Another factor is that it reduces both intracellular and extracellular PCSK9 levels.

Anti-PCSK9 siRNA and vaccine mechanisms.
Inclisiran has been modified from its precursor, ALN-PCS, to include the carbohydrate N-acetylgalactosamine (GalNAc). The addition of GalNAc allows inclisiran to be absorbed directly into hepatocytes. 48 It also boosts clinical efficacy by increasing adhesion to the hepatocytic cellular membrane. 49 Inclisiran is more convenient to administer than mAbs and only requires 2-3 doses per year. Minimal dosing also increases the chances of adherence to long-term treatment. However, it is substantially more expensive than monoclonal antibodies. This is its main drawback to date. 48
The phase 2 ORION-1 trial of inclisiran demonstrated a dose-dependent reduction of LDL cholesterol with long-lasting effects. LDL-C levels were found to be between 44.5% and 50.5% below baseline by day 30 after a single subcutaneous injection. At day 240, LDL-C levels remained 28%-36% below baseline. A second dose of inclisiran at day 90 resulted in a 41%-54% reduction at day 120 with day 240 reductions between 26% and 47%. 46 The ORION-10 and ORION-11 trials administered subcutaneous injections at days 1, 90 and every 6 months thereafter with mean reductions in LDL-C of 52% and 50% at day 510, respectively. Adverse events were similar between inclisiran and placebo groups with the exception of injection site reactions, which were more common with inclisiran (2.6% vs. 0.9% in the ORION-10 study and 4.7% vs. 0.5% in ORION-11). 50 Inclisiran is an effective and safe addition to PCSK9 inhibitors currently on the market. If production costs are reduced, it could provide a feasible option for long-term lipid management.
The siRNA inclisiran inhibits the translation of PCSK9 by degrading its messenger RNA. Inclisiran’s guide strand is complementary to the PCSK9 mRNA. The RISC complex essentially unzips the double-stranded siRNA, allowing for base pairing to take place. It then activates RNase, which cleaves the mRNA. Vaccines are designed with antigenic fragments which elicit an immune response in the host. In this case, the antibodies produced physically block PCSK9 from binding to the LDLR.
Small Molecules
The quest to utilize small molecules is the most varied method of PCSK9 inhibition. However, small molecules are hindered by PCSK9’s unique qualities and have thus been last in the race to therapeutically inhibit PCSK9. Many enzymes can be repressed by a small molecule, which competitively inhibits the active site, i.e. statins and HMGR. However, the protein-protein interaction between LDLR’s EGF-A domain and PCSK9’s catalytic domain occurs over a relatively planar surface of 530 Å2, composed of a central hydrophobic patch with salt bridges and polar interactions along the perimeter. 10 Because catalytic activity is not required for PCSK9 mediated degradation of LDLR, small molecules which inhibit its active site are restricted to the endoplasmic reticulum, where the autocatalytic processing takes place. 51,52 Nonetheless, small molecules continue to be appealing due to their low relative cost and oral administration.
There are a multitude of small molecule PCSK9 inhibitors in development. Pfizer has developed a truncated 26-residue mimetic protein, EGF-A9, which bears a resemblance to the extracellular EGF-A domain of LDLR. EGF-A9 has a twofold increased binding affinity for PCSK9 relative to wild type EGF-A (0.6 µM vs. 1.2 µM), suggesting that this could be used as a springboard for further development. 53 Genentech utilized phage display to develop a second independent EGF-A-like protein, Pep2-8, composed of 13 amino acids. With a KD of 0.7 µM, Pep2-8 is slightly inferior in binding affinity to Pfizer’s peptide. However, its small size makes it a better candidate for oral administration. 54
LIB003 is an interesting new small molecule PCSK9 inhibitor with impressive clinical results. It is a recombinant fusion protein that combines a PCSK9 binding domain (adnectin) with a human serum albumin. In a 12-week double-blind phase 2 trial, 300 mg of LIB003 reduced LDL-C by >70%. Free PCSK9 was simultaneously decreased by 88%. A follow-up 52-week open-label extension trial aimed at determining the long-term efficacy and safety of LIB003 concluded that it is well tolerated, with only mild adverse events unrelated to the compound in question. All participants were on maximally tolerated statins at the time of enrollment. The addition of LIB003 resulted in a stable reduction of LDL-C by 57%-59%. LIB003 is administered as monthly subcutaneous injections and is also stable at room temperature for up to 9 months. 55 This could reduce production and transportation costs, making it more accessible and cheaper for patients.
Other small molecules currently being investigated include MG132, 7030B-C5, PF-06446846 derivatives, and R-IMPP. 56 The proteasome inhibitor MG132 produces a dose-dependent increase in LDLR mRNA in HepG2 cells of up to 70% from initial levels. It also simultaneously decreases PCSK9 mRNA levels by 70% after 24 hours. The addition of pravastatin showed further enhancement of LDLR mRNA with a continued decrease in PCSK9 levels. 57 7030B-C5 was found to inhibit PCSK9 transcription via modulation of the transcriptional regulators HNF1α and FoxO3. In HepG2 cells, PCSK9 levels were decreased by greater than 50% with corresponding increases in LDLR expression. 12 weeks of treatment with 7030B-C5 in ApoE knockout mice on a high fat diet decreased TC and LDL-C by 15%. Atherosclerotic plaque, atherosclerotic lesions, triglycerides and glucose levels also decreased. 58 Although modest, this decrease illuminates a new method in targeting PCSK9 inhibition. PF-06446846 was found to selectively inhibit PCSK9 translation, resulting in a 30% decrease in total plasma cholesterol and 58% decrease in LDL cholesterol in rats. 59 However, its excessive toxicity has led to investigations of similar compounds with greater tolerability. 60,61 Yet another molecule in development was found to bind directly to PCSK9 and restore LDL uptake in HepG2 liver cells with the gain-of-function mutation PCSK9 D374Y. 62
Vaccines
Recently, anti-PCSK9 vaccines have made their way to the forefront of cutting edge PCSK9 research. Peptide-based vaccines work by preventing PCSK9 from binding to LDLR 63,64 (Figure 3) and in some cases also cause PCSK9 degradation. 64 Numerous murine studies have been published with impressive results. The peptide-based AT04A vaccine showed a sustained decrease in PCSK9 levels (≥49%) over 12 weeks. 65 The peptides in AT04A mimic the N-terminus of PCSK9, which is responsible for LRLR interaction. 63 After 18 weeks, PCSK9 levels remained 24% below baseline and total cholesterol was measured to be 53% less than control mice. Vascular inflammation, atherosclerotic lesions and inflammatory biomarkers were also significantly reduced. 65 L-IFPTA+ is a vaccine which incorporates an immunogenic peptide onto the surface of negatively charged nanoliposomes along with the adjuvant Alhydrogel®. Biweekly injections of L-IFPTA+ decreased PCSK9 plasma concentrations by up to 58.5% in C57BL/6 mice. After 8 weeks, LDL-C and TC levels showed a 51.7% and 44.7% decrease compared to controls. 64 Other studies have also shown success in lowering LDL-C levels through the use of PCSK9 vaccines. 63,66 -69 The diversity of these approaches confirms that there are a multitude of ways to inhibit PCSK9 via vaccination. Although the long-lasting effects of vaccines makes them attractive, this is also a cause for caution regarding possible adverse events as serum antibody levels cannot be controlled as meticulously. 66
Suggested Recommendations for the Use of PCSK9 Inhibitors
The success of PCSK9 inhibitors has already made its way into the literature, with the American Heart Association (AHA) including them in both their 2018 Guideline for the Management of Blood Cholesterol and 2019 Guideline on the Primary Prevention of Cardiovascular Disease. The 2019 guideline discusses treatments for different severities of hypercholesterolemia, determined by LDL-C and risk levels. Statins are still recommended as a primary treatment for those with ASCVD; however, one of the new additions is the attention now given to both ezetimibe and PCSK9 inhibitors. Both ezetimibe and PCSK9 inhibitors are listed as secondary treatments that can be used in certain situations where LDL-C is not at goal level after statin therapy. When the guidelines were published, evolocumab and alirocumab were the only PCSK9 inhibitors approved for use.
Both AHA guidelines suggest similar actions for those suffering from severe primary hypercholesterolemia. 70 However, the 2019 guidelines break away from set LDL-C goals and instead introduce a system of LDL-C reduction goals based upon percent reduction. They recommend reducing LDL-C levels by 50% for high-risk patients and by at least 30% for those presenting intermediate risk. 71 Alternatively, if a patient shows a clear intolerance to statins, PCSK9 inhibitors such as alirocumab or evolocumab can be used instead, as studies show very low numbers of test patients reporting the myopathy that some patients experience with statins. 32 While the updated guidelines do stress that treatment can be different for a patient based on external factors such as age, race, or sex, they also stress that treatment with statins, ezetimibe, or PCSK9 inhibitors should occur simultaneously with the promotion of a healthy lifestyle. 70
The following recommended algorithms make use of the available guidelines for the treatment and prevention of CVD while also taking additional factors such as clinical data and risk enhancers into consideration. Included are recommendations for both primary and secondary prevention modeled after the AHA guidelines (Figures 4 and 5), Adult Treatment Panel III (ATP III) guidelines (Figure 6), and the 2019 ESC/EAS Guidelines for the Management of Dyslipidemias (Figures 7 and 8).

Suggested algorithm for the use of PCSK9 inhibitor therapy for the primary prevention of cardiovascular disease based on treatment algorithm of hyperlipidemia adopted from 2019 AHA/ACC guidelines. 71 All yellow boxes represent unchanged AHA/ACC guideline adaptations. Blue boxes indicate recommendations based on the consideration of additional lipid guidelines and the presence of additional risk enhancers.

Suggested algorithm for the use of PCSK9 inhibitor therapy for the secondary prevention of cardiovascular disease based on treatment algorithm adopted from 2018 AHA/ACC guidelines. 70 All yellow boxes represent unchanged AHA/ACC guideline adaptations. Blue boxes indicate recommendations based on the consideration of additional lipid guidelines and the presence of additional risk enhancers.

Suggested algorithm for the use of PCSK9 inhibitor therapy for the primary and secondary prevention of cardiovascular disease partially based on ATP III guidelines. 72 It is of note that ATP III guidelines make no mention of PCSK9 inhibitors because they were not on the market at the time of publication. All yellow boxes represent unchanged ATP III guideline adaptations. Blue boxes indicate recommendations based on the consideration of additional lipid guidelines and the presence of additional risk enhancers.
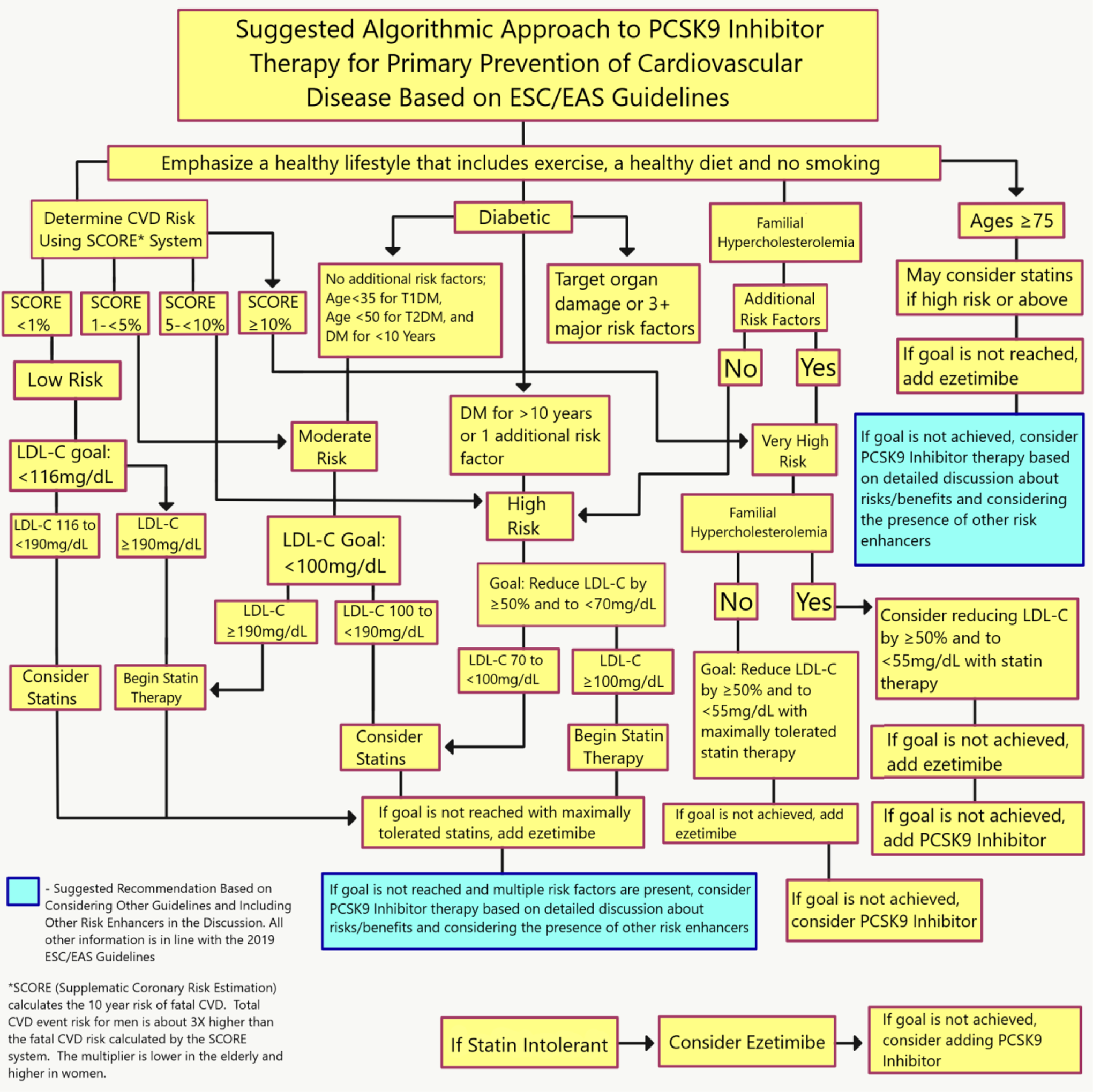
Suggested algorithm for the use of PCSK9 inhibitor therapy for the primary prevention of cardiovascular disease based on the 2019 ESC/EAS guidelines for the management of dyslipidemias. 73 All yellow boxes represent unchanged ESC/EAS guideline adaptations. Blue boxes indicate recommendations based on the consideration of additional lipid guidelines and the presence of additional risk enhancers.
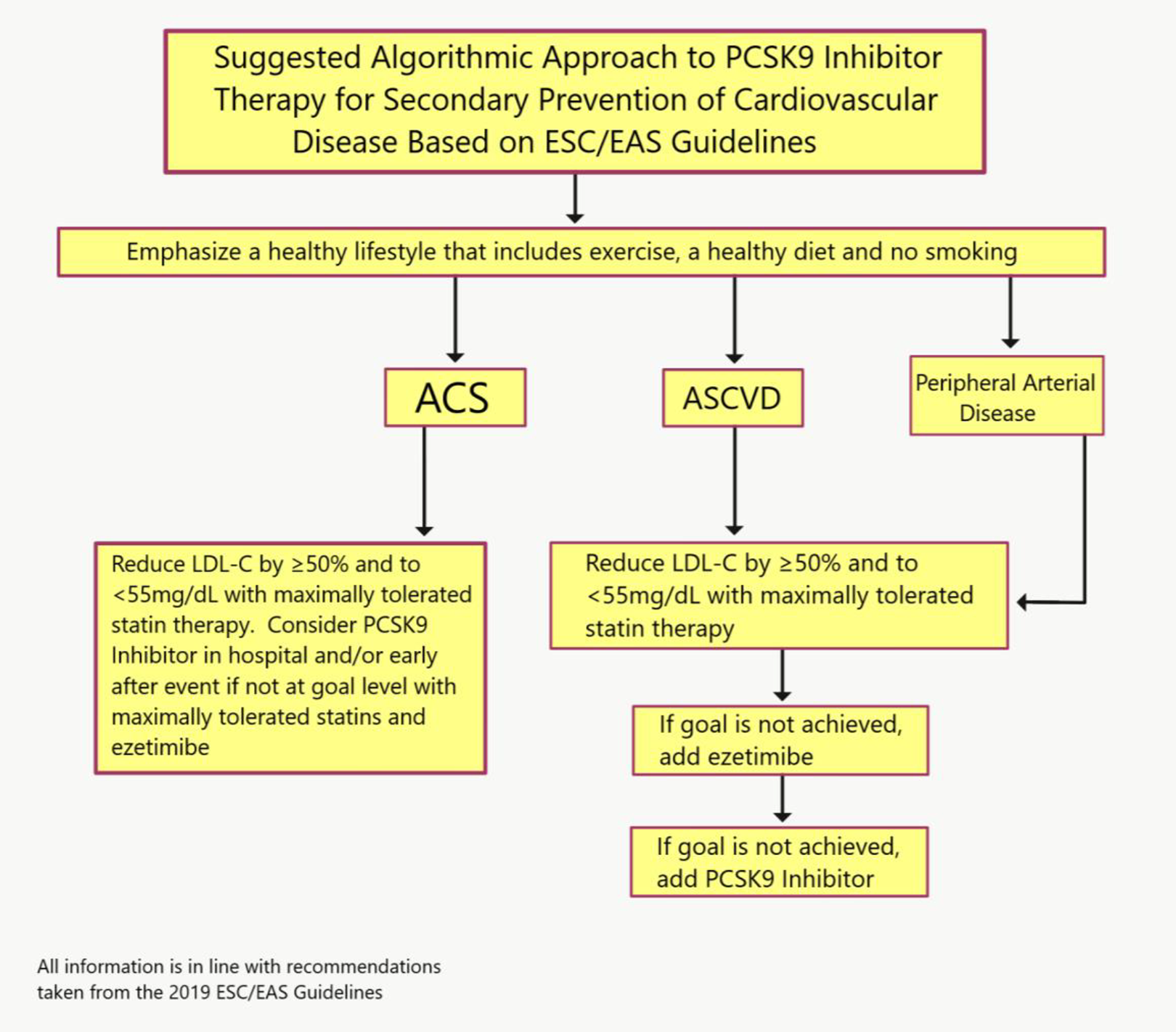
Suggested algorithm for the use of PCSK9 inhibitor therapy for the secondary prevention of cardiovascular disease based on the 2019 ESC/EAS guidelines for the management of dyslipidemias. 73 All yellow boxes represent unchanged ESC/EAS guideline adaptations.
Conclusion
PCSK9 inhibitors are a new addition to our available treatment for hyperlipidemia. Table 1 highlights basic specifications of selected PCSK9 inhibitors. In patients not at goal LDL-C levels with statins or who are statin intolerant and for those for those at risk of cardiovascular events, PCSK9 inhibitors offer a valuable addition to treatment options for high LDL-C. Limitations include the expense of these newly approved drugs and lack of availability in oral forms. Future development of orally available formulations and additional clinical trials with longer follow up will hopefully enhance utilization of this new class of drug treatment in the fight against atherosclerosis and cardiovascular disease.
Frontrunner PCSK9 Inhibitors and Their Current FDA Phase.

Footnotes
Author Contributions
All authors contributed equally to this study.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.