
Abstract
Dyslipidaemia is a critical risk factor for the development of cardiovascular complications such as ischemic heart disease and stroke. Although statins are effective anti-dyslipidemic drugs, their usage is fraught with issues such as failure of adequate lipid control in 30% of cases and intolerance in select patients. The limited potential of other alternatives such as fibrates, bile acid sequestrants and niacin has spurred the search for novel drug molecules with better efficacy and safety. CETP inhibitors such as evacetrapib and anacetrapib have shown promise in raising HDL besides LDL lowering property. Microsomal triglyceride transfer protein (MTP) inhibitors such as lomitapide and Apo CIII inhibitors such as mipomersen have recently been approved in Familial Hypercholesterolemia but experience in the non-familial setting is pretty much limited. One of the novel anti-dyslipidemic drugs which is greatly anticipated to make a mark in LDL-C control is the PCSK9 inhibitors. Some of the anti-dyslipidemic drugs which work by PCSK9 inhibition include evolocumab, alirocumab and ALN-PCS. Other approaches that are being given due consideration include farnesoid X receptor modulation and Lp-PLA2 inhibition. While it may not be an easy proposition to dismantle statins from their current position as a cholesterol reducing agent and as a drug to reduce coronary and cerebro-vascular atherosclerosis, our improved understanding of the disease and appropriate harnessing of resources using sound and robust technology could make rapid in-roads in our pursuit of the ideal anti-dyslipidemic drug.
Dyslipidemia is one of the modifiable factors that play a central role in the etiology of ischemic heart disease. The prevalence of dyslipidemia has been steadily increasing in both developed and developing countries, the latter owing to the increasing westernization of diet and lifestyle changes. According to World Health Organization estimates, more than half of the patients with ischemic heart disease have co-existing dyslipidemia. 1 Effective treatment of dyslipidemia has been shown to improve mortality and morbidity. Every 30 mg/dL decrease in low-density lipoprotein cholesterol (LDL-C) results in a 30% reduction in adverse cardiovascular events. 2 Currently, the main strategies to control dyslipidemia include statins, fibrates, bile acid sequestrants, and niacin. Statins can reduce LDL-C by 30% to 50%. However, this maximal effect can be obtained only with high doses of potent statins such as rosuvastatin and atorvastatin. This adds to the cost and reduces drug tolerability. The American Heart Association/American College of Cardiology blood cholesterol guideline that was released in November 2013 has emphasized the role of high-dose statins in patients with clinical atherosclerotic vascular disease irrespective of LDL-C levels. 3 A number of new molecules have been developed for the control of cholesterol in recent years. Studies are in progress to examine the benefit of these drugs in lipid management over and above conventional therapy and also to see whether it could provide reduction in cardiovascular events such as myocardial infarction, stroke, and mortality. Some of the earlier molecules have failed in clinical development for various reasons (Table 1). This review is a brief description of some of the promising drugs that are in clinical development for dyslipidemia.
Molecules That Have Failed in Dyslipidemia Drug Development.
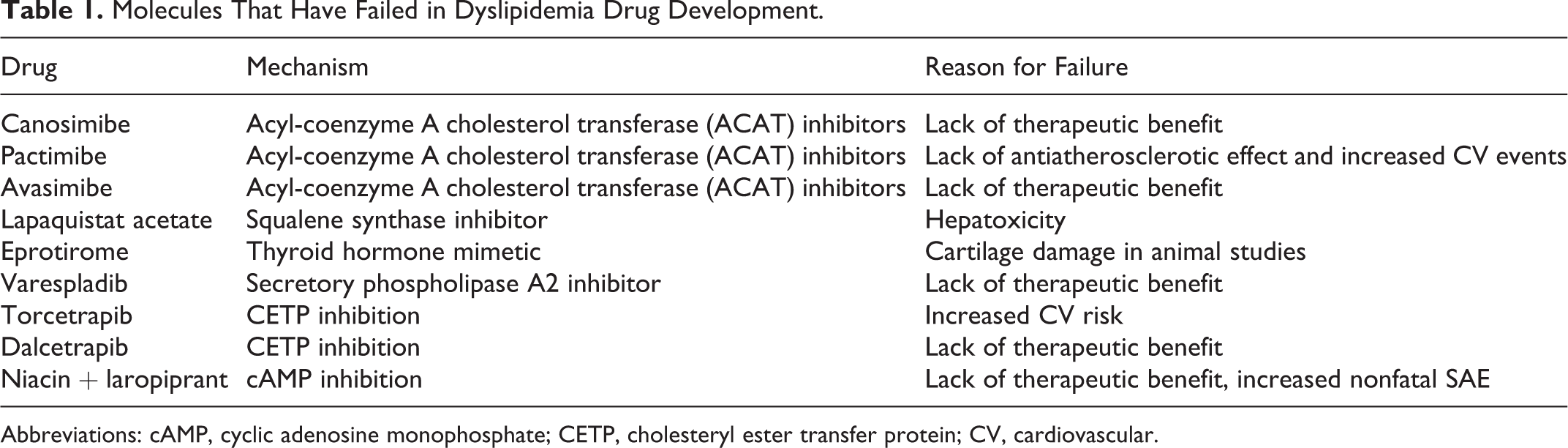
Abbreviations: cAMP, cyclic adenosine monophosphate; CETP, cholesteryl ester transfer protein; CV, cardiovascular.
Cholesteryl Ester Transfer Protein Inhibitors
High-density lipoprotein (HDL), with a protective action on the vasculature, is considered an independent negative risk factor for the occurrence of atherosclerosis. Studies have shown association between lower levels of HDL and increased risk of cardiovascular events in spite of achieving LDL reduction with statins. 4,5 In addition, HDL has been shown to possess anti-inflammatory, antioxidant, and antithrombotic effects. 6,7 Hence, measures are being explored to attenuate the risk of cardiovascular diseases through augmentation of HDL concentration apart from taking measures to lower LDL levels. One of the most promising targets under development for increasing HDL cholesterol is cholesteryl ester transfer protein (CETP).
Cholesteryl ester transfer protein is a 74-kDa hydrophobic plasma glycoprotein, synthesized from liver and adipose tissue. It is released into the circulation, binds to, and metabolizes HDL. 8 In addition, CETP plays a vital role in the bidirectional transfer of cholesteryl ester in HDL for triglycerides (TGs) in apolipoprotein B (apoB) containing lipoproteins, that is, LDL and very LDL (VLDL). As a consequence, CETP inhibition results in cholesterol retention within HDL leading to reduced concentration of lipoproteins as well as decreased risk of atherosclerosis. 9,10 This is confirmed by a prospective cohort study done in patients with CETP inhibition owing to genetic variations that resulted in diminished risk of cardiovascular and cerebrovascular diseases. Further the study reported a favorable antiatherogenic lipid profile with increased longevity in such patients. 11 The growing evidence on the association between CETP and HDL have led to the discovery and development of newer molecules to raise HDL level through CETP inhibition.
Currently, the various CETP inhibitors evaluated in clinical trials include torcetrapib, dalcetrapib, anacetrapib, and evacetrapib. 12 The Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events (ILLUMINATE) trial conducted with the first CETP inhibitor, torcetrapib, demonstrated favorable effect on HDL and LDL at the end of 12 months of treatment. However, patients on torcetrapib had increased incidence of mortality and major cardiovascular events leading to termination of the study. 13 The rise in cardiovascular complications and mortality with torcetrapib were attributed to the enhanced aldosterone concentration and the resultant increase in blood pressure. Although considered a class effect initially, the increase in aldosterone levels was later found to be an off-target action of torcetrapib alone. 14,15 The newer CETP inhibitors dalcetrapib, anacetrapib, and evacetrapib have been found to lack off-target action thus confirming that rise in aldosterone levels and the resultant cardiovascular complications of torcetrapib are not class effect. 16,17 However, dalcetrapib failed to show adequate efficacy in phase 3 clinical trials and hence had to be terminated from further drug development.
Anacetrapib, given at a dose of 60 mg/kg/d for 2 weeks, was shown to inhibit CETP by 94% resulting in increased HDL level by 47% in a dyslipidemic hamster model. This was established by an increase in free unlabeled cholesterol and cholesteryl ester confirming the occurrence of reverse cholesterol transport. 18 Likewise, anacetrapib administered either as monotherapy or in combination with atorvastatin showed significant rise in HDL-C and fall in LDL-C at the end of 8 weeks of treatment in a study done in Japanese patients with dyslipidemia. 19 In determining the efficacy and tolerability of CETP inhibition with anacetrapib (DEFINE) study, anacetrapib 100 mg once daily administered along with statins was found to increase HDL-C by 138.1% and reduce LDL-C by 39.8% following 24 weeks of treatment in patients with coronary heart disease (CHD) or CHD equivalents. The study demonstrated the sustained effect of anacetrapib on lipid levels even after 12 weeks following cessation of treatment in addition to safety and tolerability at the end of 76 weeks of study. 20 However, the rise in HDL cholesterol was found to be maintained even 2 to 4 years after the last dose. The trial with anacetrapib did not show any increase in mortality or major adverse cardiovascular events. 21,22
The newer CETP inhibitors have an additional benefit of causing significant fall in LDL apart from increasing HDL. 23,24 This phenomenon was seen with evacetrapib, a newer CETP inhibitor that showed significant increase in HDL and decrease in LDL when given either as monotherapy or as combination with statins for 12 weeks. 25 Besides, evacetrapib was shown to be more potent than torcetrapib, dalcetrapib, and anacetrapib due to complete inhibition of CETP. 16 These developments have rekindled the anticipation toward the exploration of a promising CETP inhibitor for the management of dyslipidemia and atherosclerosis. 26 Nonetheless so far, there has been lack of evidence regarding the positive consequences of increasing HDL concentration on the cardiovascular outcomes. Results of the ongoing studies are awaited to predict the potential of HDL—modifying therapies in the prevention of cardiovascular diseases. Table 2 gives a summary of the current status of CETP inhibitors.
Cholesteryl Ester Transfer Protein Inhibitors at a Glance.

Abbreviations: HDL, high-density lipoprotein; LDL, low-density lipoprotein; Lp(a), lipoprotein(a).
Microsomal TG Transfer Protein Inhibitors
Microsomal triglyceride transfer protein (MTP), a major heterodimeric lipid transfer protein, is involved in the synthesis of chylomicrons and VLDL in the intestine and liver, respectively. It is located in the endoplasmic reticulum (ER) of enterocytes and hepatocytes. Microsomal triglyceride transfer protein plays a vital role in the assembling of chylomicron with apoB48 in the intestine and VLDL with apoB100 in liver. Owing to its action on the synthesis of LDL, VLDL, apoB, and TGs, inhibition of MTP is being explored as one of the prospective targets in the treatment of hyperlipidemia. 27 In addition to having favorable effect on lowering lipid levels, MTP inhibitors are also shown to possess antiatherosclerotic and insulin-sensitizing actions. 28 These findings suggest an indirect role for MTP inhibitors in the treatment of diabetes mellitus, which has an association with enhanced apoB expression and hypertriglyceridemia. In a study done with MTP inhibitor, significant reduction in dyslipidemia related to insulin resistance with subsequent decrease in the risk of atherosclerosis was demonstrated in Zucker fatty rats. 29 The various MTP inhibitors explored so far include implitapide, dirlotapide, mitratapide, lomitapide, JTT-130, and SLX-4090. Of these, lomitapide and SLX-4090 are in clinical trials with hopeful prospect. 30 Implitapide, an MTP inhibitor, was found to inhibit atherosclerosis in apoE knockout (apoE KO) mice and was found to cause fall in both total cholesterol and TG levels following 8 weeks of treatment in apoE KO mice fed on western-type diet. 31
Animal study done in LDL receptor-deficient mice fed on western diet for 16 weeks followed by changeover to either chow diet or chow diet with MTP inhibitor, BMS 212122, for 2 weeks showed rapid reduction in plasma lipid levels. The study found a significant decrease in monocyte-derived (CD 68+) cells in atherosclerotic plaques along with increased collagen content indicating the improved stability of plaques in the MTP inhibitor-treated group compared to the control groups. Further, atherosclerotic plaques of these mice were shown to have significant decrease in proinflammatory M1 macrophage markers along with an increase in anti-inflammatory M2 macrophage markers. This study confirms the favorable effect of MTP inhibitors on the regression of atherosclerotic plaques. 32 Likewise, MTP inhibition in mice fed with western diet had been shown to cause reduction in plasma TG and cholesterol levels. However, MTP inhibitor-treated mice were found to have increased plasma alanine aminotransferase (ALT)/aspartate aminotransferases, hepatic TG, and free cholesterol levels. This rise was attributed to the occurrence of oxidative stress secondary to accumulated free cholesterol in the ER and mitochondria. 33
Among the MTP inhibitors, those selective for hepatocytes have been found to reduce VLDL secretion containing apoB100 while those selective for enterocytes have been shown to decrease secretion of chylomicrons made of apoB48. A newer candidate, namely JTT-130, with selective inhibition of intestinal MTP following oral administration was found to enhance carbon-14 radioactivity in the intestine of rats. Similarly, in hyperlipidemic hamsters fed on high-cholesterol diet, JTT-130 given for 2 weeks was found to reduce TG levels in both plasma and liver without increasing liver transaminases. The lack of hepatotoxicity with JTT-130 was attributed to the rapid degradation of the drug during the process of absorption. According to this study results, JTT-130, an intestine-specific MTP inhibitor, could emerge as a promising agent in the treatment of hyperlipidemia without causing hepatotoxicity. 34 Similarly SLx-4090, an oral MTP inhibitor with less systemic absorption, was shown to inhibit chylomicrons synthesis with selective action in enterocytes while lacking hepatotoxicity. 35
One of the MTP inhibitors, lomitapide, is emerging as a newer alternative for hypercholesterolemia in patients intolerant to conventional lipid-lowering therapies. 36,37 A phase III trial in adults with homozygous familial hypercholesterolemia (FH) showed reduction in mean plasma LDL-C by 50% from baseline at the end of 26 weeks and the effect was found to last till 78 weeks of treatment with lomitapide. But the drug was found to cause increase in the incidence of gastrointestinal adverse events and accumulation of fat in the liver. Yet the increased fat content in the liver was found to stabilize following initial rise with reversal of elevated ALT subsequent to dose reduction. 37,38 Chronic use of lomitapide was found to lack QTc interval prolonging action. 39 Based on its efficacy and safety, lomitapide has been approved for the treatment of homozygous FH recently. 40 Apart from homozygous FH, MTP inhibitors might also be appropriate in the treatment of metabolic syndrome, type 2 diabetes mellitus, familial combined hyperlipidemia, and heterozygous FH. 41 However, the safety concerns especially with respect to the occurrence of fatty liver needs to be addressed prior to the recommendation of MTP inhibitors in the treatment of hyperlipidemia and hypercholesterolemia. Table 3 summarizes the MTP inhibitors at a glance.
Microsomal Triglyceride Transfer Protein (MTP) Inhibitors at a Glance.

Abbreviations: EMA, European Medicines Agency; FDA, Food and Drug Administration; FH, familial hypercholesterolemia.
Proprotein Convertase Subtilisin/Kexin 9 Inhibitors
Proprotein convertase subtilisin/kexin 9 (PCSK9) is a serine protease involved in the degradation of the LDL receptor. It binds to the LDL receptor present on the surface of the cell via clathrin-coated pits and causes internalization of LDL receptor leading to degradation and resultant increase in concentration of LDL cholesterol. 42 A specific mutation in PCSK9 gene namely D374Y was found to cause gain of function resulting in increased binding of PCSK9 to the LDL receptor and resultant degradation. The findings related to inhibition of PCSK9 are burgeoning as an attractive target for reducing LDL-C in both dyslipidemia and cardiovascular diseases. 43,44
Monoclonal antibodies against PCSK9 have opened a newer paradigm in the reduction of LDL-C and have been found to be effective in the treatment of both familial and non-FH. 45 Evolocumab (AMG 145), a fully human monoclonal immunoglobulin G2 antibody specific for human PCSK9, has been shown to prevent degradation of LDL receptor. A phase I trial, with evolocumab, was found to produce significant reduction in serum LDL-C in healthy volunteers, hypercholesterolemic statin-treated patients, patients with heterozygous FH, and those on high doses of atorvastatin or rosuvastatin. The incidence of adverse effects observed in this trial with AMG 145 was akin to placebo. 46 Similarly, LDL-C Assessment with PCSK9 Monoclonal Antibody Inhibition Combined With Statin Therapy-Thrombolysis In Myocardial Infarction 57 (LAPLACE-TIMI 57), a 12-week, randomized, double-blind, and placebo-controlled study with different dose levels of evolocumab in combination with statins, found significant reduction in mean LDL-C by 41.8% to 66.1% every 2 weeks and 41.8% to 50.3% every 4 weeks. 47 Evolocumab in combination with statins was found to significantly reduce lipoprotein(a) by 32% in patients with hypercholesterolemia. 48
Evolocumab administered either alone or with ezetimibe in a phase II, randomized, double-blinded, and placebo-controlled study resulted in significant reduction in LDL-C among patients with heterozygous FH. In this study, the drug was shown to be well tolerated and safer with less adverse effects. 49 In another phase II randomized, double-blinded, placebo and ezetimibe controlled study done with evolocumab in patients intolerant to statin demonstrated a significant reduction in LDL-C following 12 weeks of therapy. This study came across 4 serious adverse events with evolocumab in the form of coronary artery disease, acute pancreatitis, hip fracture, and syncope. 50
Another PCSK9 inhibitor namely alirocumab (SAR236553/REGN727) given in combination with atorvastatin was found to reduce LDL-C by 40% to 72% in patients with primary hypercholesterolemia at the end of 12 weeks in a randomized, double-blind, parallel group, placebo-controlled trial. 51 Similarly in a phase II controlled trial, alirocumab given at various doses and dosing intervals along with statin was found to be well tolerated and resulted in LDL reduction among patients with heterozygous FH at the end of 12 weeks of treatment. 52 Likewise, a phase II, double-blind, placebo-controlled, randomized trial done in patients with primary hypercholesterolemia found that addition of alirocumab to atorvastatin resulted in significant reduction in LDL compared to atorvastatin alone. 53 So far, the results of PCSK9 inhibitors from various clinical trials are assuring the possibility of potential new drugs emerging against hyperlipidemia in the near future.
ALN-PCS is a small-interfering RNA (siRNA) that activates sequence specific messenger RNA for PCSK9 and thus reduces the production of PCSK9, as evidenced from animal models. This brings about a fall in the LDL-C levels due to increased LDL receptors on the hepatocyte membrane. A phase 1 trial with ALN-PCS in 32 healthy volunteers demonstrated a 40% reduction in the concentration of LDL-C. The most common adverse effect seen in half of the patients was a self-limiting rash. The drug was found to be reasonably safe. Thus, siRNA may be another viable alternative in blocking PCSK9 synthesis to achieve LDL-C control. 54,55 However, the success of this novel drug target will depend largely on the findings of the ongoing larger phase III clinical trials.
Apolipoprotein CIII Inhibitor
Apolipoprotein CIII, a proinflammatory protein dwelling on the surface of lipoproteins, causes inhibition of lipoprotein lipase as well as the uptake of TG-rich lipoprotein remnants by hepatic lipoprotein receptors. The rise in the concentration of apoCIII is associated with higher concentration of TGs thus leading to increased risk of atherosclerosis. 56 This was further confirmed by a study showing association of increased risk of CHD with LDL containing apoCIII than of LDL without apoCIII. 57 Moreover, apoCIII has been found to be associated with the occurrence of metabolic syndrome as well as type 2 diabetes mellitus due to its atherogenic action. 58 Currently, targeting apoCIII is emerging as a novel approach for the treatment of dyslipidemia, CHD, and type 2 diabetes mellitus.
Inhibition of apoCIII by antisense oligonucleotide (ASO) was shown to reduce TG level in both preclinical and clinical studies, This ensures the potential of ASO against apoCIII in the treatment of coronary artery disease. 59 Mipomersen offers a promising approach to control lipoproteins. A phase III trial with mipomersen, a second-generation ASO developed to inhibit the synthesis of apoB100 in the liver, has shown significant fall in plasma LDL-C and other apoB-containing lipoproteins involved in atherosclerosis. 60,61
Currently, mipomersen is being investigated in mild to severe FH either as monotherapy or as add-on therapy to the standard lipid-lowering drugs in patients who are unresponsive to high-dose statins. 62 A phase III trial with mipomersen 200 mg/wk administered subcutaneously in patients with FH has demonstrated a fall in LDL-C by 25% to 47% and TGs by nearly 10% without altering HDL-C levels. The adverse effects reported with mipomersen include injection site reactions, cephalgia, nasopharyngitis, myalgia, nausea, fatigue, and flu-like symptoms. Among these, injection site reactions were seen in nearly 70% to 100% of patients. Akin to MTP inhibitors, mipomersen also causes elevated liver transaminases and hepatic steatosis. 63 Similarly, mipomersen has been found to be absorbed rapidly and extensively at a dose of 50 to 400 mg/wk, with dose-dependent reduction in LDL-C. Mipomersen is excreted renally with an elimination half-life of nearly 30 days. 64,65 Based on the findings, mipomersen has been approved by US Food and Drug Administration as an adjunct therapy for the treatment of homozygous FH with an orphan drug status. 66 In contrast, the European Medicines Agency has withheld approval of the drug citing safety concerns. 67 Thus, mipomersen and the newer ASOs under clinical investigation need to go a long way before proving their mettle and safety in the treatment of dyslipidemia.
Farnesoid X Receptor Modulation
Farnesoid X receptor (FXR) also known as NR1H4 is a ligand-activated nuclear hormone receptor, which is highly expressed in liver and intestine. 68 It serves as a bile acid sensor and protects the cells from bile acid toxicity. It is primarily activated by bile acids, which is the end product of cholesterol catabolism. Activation of FXR results in downregulation of bile acid synthesis, reduced import of bile acids from the plasma into the hepatocyte, increased export of bile acids into bile, and reduced TG levels. 69 Chenodeoxycholic acid is a natural FXR agonist, which can reduce TG levels. 70 Synthetic FXR agonists such as GW4064 have been shown to reduce TG levels.
Guggulsterone is the active ingredient in guggul, an Indian shrub. Guggulsterone, which was found to have FXR antagonistic property, did not have any LDL lowering effects as expected, in a placebo-controlled clinical trial. It is well possible that modulation of FXR by other agents could result in LDL reduction, and the search is still for one such molecule. A study performed in a wild-type mice fed with high-cholesterol diet and treated with guggulsterone showed decreased hepatic cholesterol content, whereas FXR-deficient mice did not show similar effect. 71 So this shows that the hypolipemic properties of guggulsterone are mainly due to FXR antagonism.
In an animal study by Evans et al, FXR agonist WAY-362450 resulted in a significant reduction in serum TG levels than that achieved earlier by fenofibrate. There was also a consistent reduction in serum cholesterol levels in all species. 72 In summary, FXR agonist may be an interesting approach for patients with dyslipidemia. Further studies are needed to provide more information regarding the efficacy and safety of FXR agonist in humans.
Lipoprotein-Associated Phospholipase A2 Inhibitor
Lipoprotein-associated phospholipase A2 (LpPLA2) is a vascular-specific inflammatory marker that plays a regulatory role in lipid metabolism. 73 The plaque vulnerability occurs due to release of small molecules by LpPLA2, which stimulate macrophage recruitment and evolve to foam cells. The LpPLA2 has proinflammatory properties that ultimately lead to the progression of atherosclerosis. 74 The action of this enzyme is to catalyze the hydrolysis of oxidized phospholipids on LDL to lysophospholipid and oxidized fatty acids in the atherosclerotic plaque. It has been found that serum LpPLA2 level can predict the vulnerability of the coronary atherosclerotic plaque. 75
Earlier studies have shown an association between raised levels of LpPLA2 and increased cardiovascular events. 76 A meta-analysis conducted with individual records from 79 036 participants in 32 prospective studies showed that LpPLA2 activity and mass levels were associated with enhanced risk of CAD that was similar in magnitude to blood pressure and non-HDL cholesterol. 77
Darapladib is an orally active inhibitor of LpPLA2 recently developed by GlaxoSmithKline. The half-life is approximately 126 hours, and the drug’s pharmacokinetics support once-daily clinical dosing. The mechanism of darapladib is to reduce lysophosphatidylcholine content and thereby bring about a reduction in the plaque and necrotic core area. Darapladib significantly reduced lesion LpPLA2 activity and its product lyso-PC in a diabetic–hypercholesterolemic swine model. 74
Two phase 2 clinical trials have been performed with darapladib so far. One study evaluated the effects of darapladib in 959 patients with CAD or CAD risk equivalent who were earlier randomized to atorvastatin 20 or 80 mg and then randomized to oral darapladib 40, 80 160 mg, or placebo for 12 weeks. It concluded that the overall dose-dependent inhibition of LpPLA2 activity was sustained during the study and was seen in both atorvastatin dose groups, at different baseline LDL cholesterol (LDL-C; < or ≥70 mg/dL) and HDL cholesterol (HDL-C) < or ≥40 mg/dL. 78
Another study named Integrated Biomarkers and Imaging Study 2 (IBIS-2) recruited 330 patients with acute coronary syndrome or chronic coronary artery disease. Patients were randomized to receive darapladib (160 mg) or placebo. Darapladib was found to significantly inhibit plasma LpPLA2 activity in a dose-dependent manner and also prevent necrotic core expansion of coronary plaque when measured by intravascular ultrasound. 79 No major safety concerns was identified with darapladib. An unpleasant body odor is a peculiar side effect seen in some patients, which resolves spontaneously.
Two large phase 3 studies with darapladib were planned to study the effect of darapladib on cardiovascular outcomes in patients with stable coronary artery disease. One is the STABILITY study, a randomized, placebo-controlled, double-blind, parallel group, international multicenter, event-driven trial of 15 828 patients with chronic CHD receiving standard of care to darapladib enteric-coated tablets, 160 mg or placebo. The results of the study were however disappointing as darapladib did not significantly reduce the risk of the primary composite end point of cardiovascular death, myocardial infarction, or stroke. 80 Another study is the SOLID-TIMI 52, a randomized, double-blind, placebo-controlled, multicenter trial to assess the efficacy and safety of darapladib as compared with placebo in patients after an ACS. 81 The results of this study were no different, with darapladib being unable to reduce the risk of cardiovascular deaths, MI, or revascularizations compared to placebo. 82 The main side effects found with this drug include an unpleasant body odor in some patients, which usually settles with continued use. Nevertheless, the absence of favorable outcomes in the primary end points of these 2 large multicentric clinical trials makes it less likely for darapladib to be approved by the regulatory agencies in the near future.
Bromo and Extraterminal Protein Bromodomain Inhibitor
Bromodomains are protein interaction modules that serve as transcriptional regulators, chromatin modulators, and chromatin-modifying enzymes. The bromo and extraterminal proteins (BETs) are nodal transcriptional regulators that participate in cellular proliferation, cell cycle progression, and apoptosis. RVX-208 is a compound derived from resveratrol that is a BET bromodomain inhibitor. Inhibition of BET bromodomains is associated with apoA1 upregulation and increasing HDL levels. These functional HDL particles can help in retarding atherosclerosis by removing plaques by reverse cholesterol transport from the walls of the arteries and subsequent elimination by the liver. 83 The drug is the first BET antagonist that has been tested in the clinic and a phase 2 clinical trial, SUSTAIN has shown satisfactory results in the primary end point, which was improvement in HDL levels. The ASSURE trial, which was another phase 2 clinical trial, did not meet its primary end point of reduced plaque volume. However, the other secondary end points such as total atheroma volume, improvement in HDL and apoA1 were met with statistical significance. 84
Figure 1 is an overview of the mechanisms of action of the different upcoming drugs for dyslipidemia. Although there is a strong case for the use of high-dose statins in lipid control and cardiovascular prevention, the intolerance seen in some patients with high-dose statin therapy coupled with failure of lipid control gives a definite incentive to develop new molecules with better safety profile and improved cardiovascular outcomes. Although several drug failures have occurred in the past, the novel targets that are presently being harnessed may give room for optimism in the days to come.

Emerging antidyslipidemic drugs at a glance. Dashed arrow indicates inhibitory action. CETP indicates cholesteryl ester transfer protein; MTP, microsomal triglyceride transfer protein inhibitors; PCSK9, proprotein convertase subtilisin/kexin 9; apoCIII, apolipoprotein CIII inhibitor; FXR, farnesoid X receptor; LpPLA2, lipoprotein-associated phospholipase A2; LDL, low-density lipoprotein; LDL-R, LDL receptor; CE, cholesteryl ester; LDL-CE, low-density lipoprotein cholesteryl ester; HDL-CE, high-density lipoprotein cholesteryl ester; VLDL-CE, very low-density lipoprotein cholesteryl ester; TGs, triglycerides; ACAT, acetyl-coenzyme A acetyl transferase; LCAT, lecithin cholesterol acyl transferase; Lyso-PC, lysophospholipid; oxidized FA, oxidized fatty acid.
Conclusion
Dyslipidemia remains a critical risk factor in the pathophysiology of ischemic heart disease. More than half of the patients do not achieve adequate LDL-C, despite maximum dosing with statins. Novel drug targets that have evoked great attention include CETP, PCSK9, apoCIII, LpPLA2, and MTP. Cholesteryl ester transfer protein inhibitors such as evacetrapib and anacetrapib show promise and are in late clinical development. Mipomersen, an ASO agonist, and lomitapide are drugs, which have been found to have greater utility in FH. The PCSK9 inhibitors have attracted a great deal of attention owing to substantial reduction in LDL control over and above that seen with statin therapy, and results from larger trials are being eagerly anticipated. Two monoclonal antibodies evolocumab and alirocumab and a siRNA molecule ALN-PCS have been developed to inhibit PCSK9. Among the upcoming molecules, they appear to show the greatest promise in terms of cholesterol reduction. It is crucial to know whether these molecules could offer additional advantages of reducing cardiovascular mortality and LDL control over and above that offered by statin therapy.
Footnotes
Author Contributions
George, Selvarajan, Muthukumar, and Shanmugam contributed to conception and design; contributed to acquisition, analysis, and interpretation; drafted the article, critically revised the article, gave final approval, and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.