
Abstract
Clinical trials and meta-analyses have shown that statins can dose dependently increase the incidence of new-onset diabetes mellitus (DM) especially in patients with underlying abnormalities of carbohydrate homeostasis. Mendelian randomization studies support these findings since genetic variants in the gene encoding the target of statins, the enzyme 3-hydroxy-3-methylglutaryl coenzyme A reductase, are associated with increased incidence of new-onset DM, suggesting that the so-called diabetogenic effect of statins is an “on-target effect” possibly related to their main mechanism of action, that is the increased low-density lipoprotein (LDL) receptor expression. Additionally, Mendelian randomization studies have shown that genetic variants as proxies of other drugs that increase LDL receptor expression (ezetimibe and proprotein convertase subtilisin/kexin type 9 [PCSK9] inhibitors) also increase the risk of new-onset DM. This concept is supported by the fact of decreased DM prevalence in patients with familial hypercholesterolemia who have decreased LDL receptor expression. In contrast, hypolipidemic drugs, such as the cholesteryl ester transfer protein inhibitors, that decrease LDL cholesterol without directly interfering with the LDL receptor expression do not seem to detrimentally affect carbohydrate homeostasis. However, the clinical trials of ezetimibe and PCSK9 inhibitors have not shown an increased DM risk, possibly suggesting that other potential non-well-defined “off-target effects” of hypolipidemic drugs may affect carbohydrate homeostasis. Thus, the long-term effect of hypolipidemic drugs on DM risk depends not only on their final mechanism of hypolipidemic action but also on other “on-target” and “off-target” effects of these drugs.
Clinical trials and meta-analyses have clearly shown that statins can increase the incidence of new-onset diabetes mellitus (DM). 1 –3 This increased DM risk is dose dependent and is mainly observed in patients with underlying abnormalities of carbohydrate homeostasis such as prediabetes. 2,4 –7
Mendelian randomization studies have reconfirmed these findings since genetic variants in the gene encoding the target of statins, the enzyme 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), are not only associated with decreased low-density lipoprotein (LDL) cholesterol concentration and cardiovascular risk but also with increased incidence of new-onset DM (Table 1A). 8 –10 Thus, Mendelian randomization studies suggest that the so-called diabetogenic effect of statins is an “on-target effect” possibly related to their main mechanism of action, that is, the inhibition of the HMGCR, the reduction in intracellular cholesterol levels, and subsequently the increased LDL receptor expression. 9 Even though different mechanisms leading to decreased insulin sensitivity and insulin secretion by the pancreatic β cells have been implicated, 12,13 this “on-target effect” of statins implies the significance of the increased cholesterol content in the pancreatic β cells (Figure 1). In fact, experimental studies confirmed the deleterious effect of intracellular cholesterol on pancreatic β-cell function. 14 –16

Hypolipidemic drugs, LDL receptor expression, and diabetes mellitus. LDL indicates low-density lipoprotein; PCSK9, proprotein convertase subtilisin/kexin type 9.
Hypolipidemic Drugs and DM: The Results of the Mendelian Randomization Studies.

Abbreviations: CAD, coronary artery disease; CHD, coronary heart disease; CI, confidence interval; DM, diabetes mellitus; HMGCR, enzyme 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase; LDL, low-density lipoprotein; MI, myocardial infarction; NPC1L1, Niemann-Pick C1-like 1; PCSK9, proprotein convertase subtilisin/kexin type 9.
Interestingly, even though clinical trials as well as the results of the long-term IMProved Reduction of Outcomes: Vytorin Efficacy International Trial did not show an increased DM risk with ezetimibe administration, 17,18 1 Mendelian randomization study showed an increased risk of type 2 DM in individuals carrying genetic variants of Niemann-Pick C1-like 1 protein, the target of ezetimibe (Table 1B). 8 These findings are not unexpected since ezetimibe in turn can also increase LDL receptor expression. However, a recent genetic study did not support the deleterious effects of ezetimibe on carbohydrate homeostasis (Table 2). 19
Mendelian Randomization Analyses (as Proxy of Hypolipidemic Drugs Action) and Risk of Dysglycemia. 19,a

Abbreviations: ALDH2, aldehyde dehydrogonase 2; ApoB, apolipoprotein B; CAD, coronary artery disease; CETP, cholesteryl ester transfer protein; CI, confidence interval; DM, diabetes mellitus; HMGCR, enzyme 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase; LpA, lipoprotein (a); LDL, low-density lipoprotein; NPC1L1, Niemann-Pick C1-like 1 protein; PCSK9, proprotein convertase subtilisin/kexin type 9; PLG, plasminogen; SD, standard deviation.
aThe data presented above indicate that (1) the positive association of LDL-C with CAD is independent of the negative association of LDL-C with DM, (2) statin-associated loci are related to glycemia, (3) loci that encode targets of nonstatin LDL-C lowering drugs are not related to dysglycemia.
Furthermore, even though in the Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk trial no increased risk of new-onset DM was observed with evolocumab administration, 20,21 in accordance with the results of clinical trials of short duration with both evolocumab and alirocumab, 22,23 3 Mendelian randomization trials have clearly shown that genetic variants of proprotein convertase subtilisin/kexin type 9 (PCSK9) as proxies of drug-associated PCSK9 inhibition are associated with an increased risk of new-onset DM (Table 1C), 9 –11 a finding which also is not supported by a recently published study (Table 2). 19
It is worth mentioning that all drugs associated with a detrimental effect on carbohydrate homeostasis (statins, ezetimibe, PCSK9 inhibitors) share a common mechanism of hypolipidemic effect, that is elevated expression of LDL receptors, which is associated with an upregulation of LDL receptors in the pancreatic β cells leading to lipid accumulation and β-cell dysfunction (Figure 1). In accordance with these suggestions, it has been found that genetically induced higher circulated LDL cholesterol levels are associated with a lower risk of T2DM. 24 Furthermore, a genome-wide association study showed that genetically instrumented elevation by a standard deviation of LDL cholesterol (equivalent to 38 mg/dL) is associated with higher cardiovascular risk but lower risk of type 2 DM (odds ratio: 0.79, 95% confidence interval: 0.71-0.8). 25
The importance of increased LDL receptor-mediated detrimental effect of statins and possibly of other hypolipidemic drugs is supported by a study which focused on the prevalence of type 2 DM in patients with familial hypercholesterolemia (FH), who have decreased LDL receptor expression. 26 As shown in Table 3, the prevalence of type 2 DM in patients with FH is lower compared with their unaffected relatives. Additionally, it seems that the severity of the underlying mutation is inversely related to the prevalence of diabetes, since patients with mutations in the apolipoprotein β gene (who exhibit a less severe FH phenotype) exhibit higher prevalence of type 2 DM compared with patients with mutations in the LDL receptor gene. Furthermore, LDL receptor-negative patients with the highest LDL cholesterol levels had the lowest prevalence of type 2 DM. These findings reinforce the suggestion that the transmembrane cholesterol transport is causally related to the development of type 2 DM. 26
Familial Hypercholesterolemia (FH) and Prevalence of DM. 26
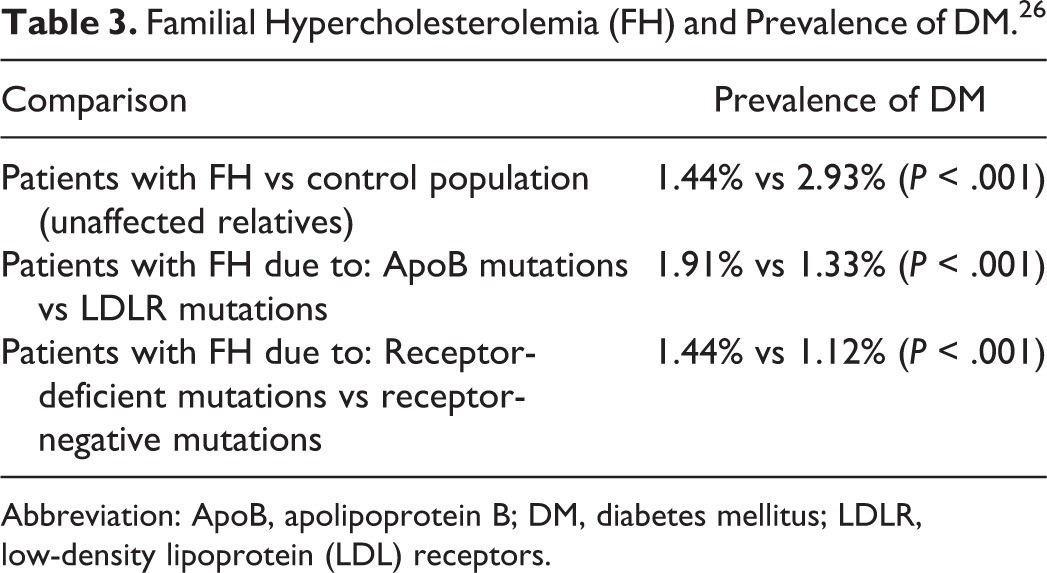
Abbreviation: ApoB, apolipoprotein B; DM, diabetes mellitus; LDLR, low-density lipoprotein (LDL) receptors.
On the other hand, this assumption is not supported by a recently published systematic review of studies focusing on patients with familial hypobetalipoproteinemia (FHBL) due to either mutations of apolipoprotein B and PCSK9 (FHBL1) or angiopoietin-like 3 (FHBL2). 27 This analysis showed that the standardized prevalence rate of DM in FHBL1, FHBL2, and the reference population is 8.2%, 4.9%, and 9.2%, respectively. Thus, low LDL cholesterol levels do not correlate with DM. This analysis, although it included a rather low number of patients with FHBL and had some limitations, reinforces the notion that other mechanisms affecting insulin resistance, insulin secretion, or both may explain the diabetogenic effects of statins. 27 According to this assumption, there may be differences among particular drugs of this class, with pravastatin and pitavastatin exhibiting a neutral or even a beneficial effect on glucose homeostasis. 28,29
Potential explanations for the different results of clinical trials and Mendelian randomization studies include the relatively short-term duration of the trials in contrast to the lifelong exposure to the natural genetic variation, as well as the potential non-well-defined “off-target effects” of drug treatment on carbohydrate homeostasis. It is worth mentioning that both HMGCR and PCSK9 genetic variations are also associated with changes in body weight and waist to hip ratio, parameters known to be related to an increased risk of new-onset DM (Figure 1). 10,11,30 It is not clear in which degree the genetic variation–mediated changes in body weight contribute to the drug-induced carbohydrate homeostasis abnormalities. However, increased body weight associated with central distribution of fat may be related to insulin resistance, which along with a decreased cholesterol-mediated insulin secretion could result in dysglycemia and type 2 DM (Figure 1).
It is worth mentioning that hypolipidemic drugs that decrease LDL cholesterol without directly interfering with the LDL receptor expression, such as cholesteryl ester transfer protein (CETP) inhibitors, 31 do not seem to detrimentally affect carbohydrate homeostasis as shown in the recently published Assessment of Clinical Effects of Cholesteryl Ester Transfer Protein Inhibition with Evacetrapib in Patients at a High Risk for Vascular Outcomes trial. 32 In fact, in the Investigation of Lipid Level management to Understand its iMpact IN ATherosclerotic Events trial, torcetrapib was followed by a beneficial effect on parameters of carbohydrate homeostasis in patients with type 2 DM, 33 and in the Randomized EValuation of the Effects of Anacetrapib Through Lipid modification trial, the incidence of new-onset DM was lower in the anacetrapib group than in the placebo group. 34 Additionally, experimental data have suggested that the dalcetrapib-associated increased cholesterol efflux from the pancreatic β cells is associated with lower intracellular cholesterol and subsequently with increased glucose-stimulated insulin secretion. 35 These results have been reconfirmed by a recently published genetic study which showed no correlation of CETP gene variants with an increased risk of type 2 DM. 19
Interestingly, the rs429937G single-nucleotide polymorphism of adenosine triphosphate-binding cassette subfamily G member 5/8 (ABCG 5/8) transporter as a proxy of bile acid sequestrants treatment is not associated with changes in parameters of carbohydrate homeostasis or in the risk of type 2 DM, 36 even though these drugs (mainly colesevelam) may beneficially affect glucose metabolism through decreased glucose absorption, increased secretion of glucagon-like peptide-1, and other mechanisms. 37,38
Thus, the long-term effect of hypolipidemic drugs on parameters of glucose homeostasis is not only related to their final mechanism of hypolipidemic action (association or not with increased LDL receptors expression) but also to other “on-target” (eg, the colesevelam-induced improvement in carbohydrate variables) and “off-target” (such as alterations in insulin sensitivity) effects of these drugs. It should be mentioned that unidentified genetic loci related to DM may exist; thus, data from genetic studies may not be conclusive. Additionally, the long-term studies that will examine the association of hypolipidemic drugs other than statins with new-onset DM are needed.
Footnotes
Authors’ Note
Filippatos contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted the manuscript, and critically revised the manuscript. Panagiotopoulou contributed to design, contributed to acquisition, analysis, and interpretation, drafted the manuscript. Tzavella contributed to acquisition, analysis, and interpretation. Elisaf contributed to conception and design and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: This editorial was conducted independently. Moses Elisaf reports personal fees from Astra-Zeneca, grants and personal fees from MSD, personal fees from Pfizer, Abbott, Sanofi-Aventis, Boehringer Ingelheim, Eli-Lilly, and GSK. Panagiotopoulou, Tzavella, and Filippatos have given talks and attended conferences sponsored by various pharmaceutical companies, including Bristol-Myers Squibb, Pfizer, Lilly, Abbott, Amgen, AstraZeneca, Novartis, Vianex, Teva, and MSD.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.