
Abstract
Diabetes is a global epidemic disease, which leads to multiorgan dysfunction, including heart disease. Diabetes results from the limited absorption of glucose into insulin-sensitive tissues. The heart is one of the main organs to utilize glucose as an energy substrate. Glucose uptake into striated muscle is regulated by a family of membrane proteins called glucose transporters (GLUTs). Although calcium channel blockers, including diltiazem, are widely prescribed drugs for cardiovascular diseases, including in patients with diabetes, their pharmacological effects on glucose metabolism are somewhat controversial. We hypothesized that diltiazem treatment will exhibit detrimental effects on whole body glucose homeostasis and glucose transport in the striated muscle of patients with diabetes. Healthy and streptozotocin-treated rats were randomly assigned to receive diltiazem treatment or a placebo for 8 weeks. Blood glucose was significantly increased in the untreated diabetic groups, which worsened after diltiazem treatment. Diabetes decreased protein content of both GLUT4 (the predominate insulin-sensitive glucose transporter) and AS160 (Akt Substrate at 160 kDa, the downstream protein in the signaling cascade that regulates GLUT4 trafficking) in striated muscle of diabetic rats, with a more pronounced alteration after diltiazem treatment. We additionally reported that diabetic rodents displayed marked systolic dysfunction, which was not rescued by diltiazem treatment. In conclusion, diltiazem treatment worsened the effects of diabetes-induced hyperglycemia and diabetes-induced alterations in the regulation of glucose transport in striated muscle.
Introduction
The incidence of both diabetes and cardiovascular diseases is anticipated to increase to epidemic levels in both the industrial and developing worlds over the next 2 decades. 1 Diabetes mellitus is a serious metabolic disorder resulting in hyperglycemia due to decreased glucose uptake in insulin-sensitive tissues, secondary to a lack of insulin production or action. Diabetes frequently leads to multiorgan dysfunction, including heart disease. Indeed, patients with diabetes are at least 10 times more likely to suffer from heart dysfunction or failure than their nondiabetic counterparts. 2 The etiologies of diabetes are complex and multifactorial, and the mechanisms underlying diabetic cardiac dysfunction remain poorly understood. Nevertheless, it is clear that impaired Ca2+ homeostasis is a hallmark feature of diabetic cardiomyopathy, 3,4 and intracellular Ca2+ overload has been reported in the heart during diabetes. 3,5 As cardiovascular disease has progressively increased worldwide, the prescription of calcium channel blockers (CCBs) has become a common place therapy as both negative inotropic and chronotropic agents, as well as class IV antiarrhythmic agents. 6 The CCBs, including the commonly prescribed nondihydropyridine CCB diltiazem, block the uptake of calcium through L-type calcium channels in the heart at therapeutic doses, thus decreasing both the rate and force of contraction. The CCBs are widely prescribed for cardiovascular diseases with 92 million prescriptions in the United States in 2009. 7 In addition, diltiazem in particular is currently the most frequently prescribed CCB for atrial fibrillation, 8 including for patients with diabetes who are more prone to arrhythmias due to diabetic complications. 9 However, the potential therapeutic beneficial effect of the nondihydropyridine on cardiac function is somewhat controversial. 10 -12
The heart is one of the main organs to utilize glucose as an energy substrate. Glucose uptake into striated muscles is regulated by a family of membrane proteins, the glucose transporters (GLUTs). 13 Glucose transporter 4 (GLUT4) is the predominately expressed transporter in these insulin-sensitive tissues and the translocation of GLUT4 from an intracellular nonactive pool to the active cell surface is the rate-limiting step in glucose transport. 14 This translocation is regulated by both insulin- and calcium-contraction-dependent processes. 15,16 These processes typically proceed via a downstream signaling pathway involving the phosphorylation of Akt Substrate of 160 kDa (AS160) that releases the intracellular GLUT4 to translocate to the active cell surface and allow the diffusion of glucose into the cell. 17 Importantly, we demonstrated that calcium is a major regulator of GLUT trafficking by an AS160-dependent pathway. 16 However, the role of abnormal Ca2+ metabolism, observed during diabetic cardiomyopathy, on glucose transport in the heart is currently poorly understood.
Interestingly, it has been reported that in vitro addition of CCBs inhibited both the insulin- and calcium-stimulated glucose transport rate in skeletal muscle, adipocytes, and neuronal cells of nondiabetic rodents. 18 -23 Conversely, Afzal et al. reported that verapamil prevented diabetes-induced myocardial changes by improving myocardial high-energy phosphate stores and ultrastructural damage without affecting overall hyperglycemia, 24 and Nagai et al. did not find unfavorable effects on glucose tolerance and insulin secretion. 25 In contrast, some physicians warn of potential adverse effects of using calcium channel antagonists in patients with diabetes, 26 and more recent epidemiological studies do not yet advocate for the prescription of CCBs for the prevention or treatment of diabetes. 27 Furthermore, hyperglycemia is a hallmark clinical finding associated with excessive CCB dosing, which is often fatal. 28 Thus, although CCBs are widely prescribed for cardiovascular diseases, data regarding their effects on glucose metabolism in diabetic and nondiabetic patients have been somewhat equivocal. This area of pharmacology is certainly deserving for further investigation, especially given the relatively high incidence of cardiovascular diseases in patients with diabetes. 29
Here, we hypothesized that long-term treatment with diltiazem will exhibit a detrimental effect on whole body glucose homeostasis and glucose transport in the striated muscle of diabetic rodents.
Methods and Materials
Animals
All procedures were approved by the Institutional Animal Care and Use Committees. Four groups of male Wistar rats were used. At 10 weeks of age, diabetes (Dx) was induced by intraperitoneal injection of a low dose of streptozotocin (STZ, 50 mg/kg in citrate buffer) in 2 groups, while the other 2 groups received a placebo injection (citrate buffer alone). 3,5 One group of diabetic (Dx-CCB) rats and one group of healthy rats (Co-CCB) were randomized to receive diltiazem (25 mg/kg/d orally) for 8 weeks. 30 The volume of diltiazem delivered in the drinking water was calculated based on the individual water consumption, which was measured every other day. Rats were housed separately to appropriately monitor individual diltiazem consumption. Diltiazem treatment was administered at the onset of STZ injections, in order to achieve adequate therapeutic range by the time animals became fully diabetic (Figure 1B). 31,32

Type 1 diabetes results in sustained hyperglycemia and weight loss. Mean ± SE (A) body weight and (B) fasting blood glucose concentration in untreated healthy (Co), diabetic (Dx), healthy diltiazem-treated (Co-CCB), and diabetic diltiazem-treated (Dx-CCB) rats, obtained at baseline (week 0) and up to 8 weeks after STZ injection. Co and CoCCB: n = 3/group; Dx: n = 11; DxCCB: n = 6. ‡P < .05 versus baseline, *P < .05 versus control, †P < .05 versus Dx. SE indicates standard error; STZ, streptozotocin.
In Vivo Experiments
Body weight and general condition of the rats were monitored weekly. Venous blood was drawn weekly from the tail vein of animals fasted overnight. Blood glucose concentration was measured using a glucometer (Bayer Contour, Tarrytown, New York). Serial echocardiographic examinations were performed at baseline and 8 weeks after injection. Two-dimensional and M-mode echocardiographic images, and Doppler recording were obtained using a GE Vivid-7 echocardiograph system (11.5 MHz sectorial probe), following standard techniques, as previously described by our group. 3,5,33 Ventricular structure and function were assessed by measuring left ventricle (LV) internal diameter (LVID) end-diastolic and end-systolic chamber dimensions and wall thickness from M-mode tracings, and LV fractional shortening (LVFS): (LVFS = [(LVIDs − LVIDd)/LVIDd] × 100). Color flow guided, pulsed-wave Doppler imaging was recorded for pulmonary artery, pulmonary venous, and mitral flows. Echocardiographic image measurements were performed offline. All image acquisitions and off-line measurements were conducted by the same investigator. Average values were obtained from the measurement of 3 cardiac cycles from one cine loop. After the completion of the 8-week period, animals were sacrificed and tissues collected for Western blotting.
Striated Muscle Protein Extraction
Cardiac (right ventricle and LV) and skeletal (soleus) muscles were collected 8 weeks after the induction of diabetes. Crude membrane extracts for analysis of GLUT4 was obtained, as previously described by our group. 34,35 Briefly, tissue samples were homogenized in buffer and protease inhibitor (Sigma, St. Louis, Missouri) and crude extracts of plasma membranes were obtained by centrifugation at 100 000× g for 90 minutes, following which the pellet was resuspended and used for analysis. 34 -36 Cardiac and skeletal muscle total tissue lysates for analysis of (total and phosphorylated) AS160 were obtained as described previously by our group. 35 Total tissue and crude membrane extracts were analyzed for total protein concentration by bicinchoninic acid assay, using a commercially available kit (Thermo Fisher Scientific, Walktham, MA) and spectrophotometer.
Western Immunoblotting
AS160 and ryanodine receptor 2 (RyR2) antibodies were purchased from Thermo Fisher. The GLUT4 and total and phospho (serine/threonine) AS160 antibodies (against human) were purchased from AbD Serotec (Raleigh, North Carolina) and Cell Signaling (Danvers, MA), respectively, and were chosen based on their sequence homology with rat proteins. All antibodies were validated against a positive control.
Crude membrane extracts (for GLUT content) and total tissue lysates (for AS160 content) were analyzed in duplicate for protein contents by electrophoresis and subsequent quantitative Western blotting, as previously described by our group. 15,35 -37 Briefly, equal amounts of protein (25-75 μg) were resolved on a 7% (AS160) or 12% (GLUTs) sodium dodecyl sulfate -polyacrylamide gel (Mini-protean II, Bio-Rad, Hercules, California) and then electrophoretically transferred to a polyvinylidene fluoride membrane (Millipore, Billerica, Massachusetts). After blocking with 5% to 10% nonfat dry milk solution, membranes were incubated with primary antibodies overnight (SERCA2a: dilution 1:1000, Thermo Fisher, Walktham, MA, #MA3-919; RyR2: dilution 1:3000, #Thermo Fisher MA3-916; total and phospho-AS160: dilution 1:1000, Cell Signaling, Danvers, MA, #2670 and #9611, respectively) or for 1 hour (GLUT4: dilution 1:7500, Bio-Rad #4760-1704). After being washed with TBS–0.1% Tween 20, the membranes were incubated with an anti-rabbit horseradish peroxidase-linked secondary antibody. Primary antibodies were chosen based on their 100% sequence homology with the protein of interest in rodents, and validated against a positive control (ie, tissue, peptide) and/or negative control, as appropriate. Quantitative determination of proteins was performed by autoradiography after revealing the antibody-bound transporter protein by enhanced chemiluminescence reaction (KPL, Gaithersburg, Maryland). The density of the bands on scanned autoradiographs was quantified relative to an internal positive control sample using a Gel-Pro analyzer blot scanning and analysis system (Media Cybernetics, Bethesda, Maryland). Equal protein loading for GLUT4, SERCA2a and RyR2 was confirmed by stripping the membranes, and subsequent reblotting with calsequestrin primary antibody (dilution 1:2500, Thermo, Waltham, MA). Equal protein loading for total and phospho-AS160 was confirmed from α-tubulin or β-actin visualized by MemCode reversible protein staining (Thermo, Waltham, MA).
Statistics
Data are presented as mean ± standard error. Differences between means were assessed using a statistical software package (Sigmaplot 11.0, Systat Software, Inc, San Jose, California). Two-way repeated analysis of variance (ANOVA; treatment and time factors) for in vivo measurements and a one-way ANOVA (treatment factors) for in vitro measurements were performed, as appropriate. When a significant difference was identified, post hoc tests were performed using the Student-Newman-Keuls test. Correlation between an independent and dependent variable was determined by regression analysis. Statistical significance was defined as P < .05.
Results
Effects of Diltiazem Treatment on In Vivo Metabolic Parameters and Cardiac Function
To explore the in vivo effects of diltiazem treatment on healthy and diabetic rodents, we measured blood glucose and body weight before, during, and after the treatment period. As expected, untreated diabetic rats became markedly hyperglycemic and lost body weight over the 8-week period, when compared both to their own baseline measurements and to matched control animals (P < .001, Figure 1). Long-term treatment with diltiazem for 8 weeks not only did not improve the hyperglycemic condition, but in fact worsened hyperglycemia 3 days following the injection of STZ (P < .001). Similarly, body weight was not significantly improved by diltiazem treatment.
Ventricular structure and function were assessed by 2-dimensional cine loops of a long-axis view and of a short-axis view at midlevel of the papillary muscles, as well as M-mode loops of the short-axis view. Ventricular fractional shortening, a surrogate of systolic dysfunction (Table 1), was decreased in untreated diabetic rats at 8 weeks after STZ injection when compared to their own baseline measurements (42.43% ± 1.21% at baseline to 35.35% ± 1.21% at 8 weeks, P < .05). In addition, end systolic volume was increased in untreated diabetic rats at 8 weeks after STZ injection (0.24 ± 0.02 mL at baseline to 0.40 ± 0.02 mL at 8 weeks). As a result, cardiac output (0.27 ± 0.02 L/min at baseline to 0.22 ± 0.02 L/min at 8 weeks) and pulmonary ejection velocity were significantly decreased. Furthermore, both treated and untreated patients with diabetes developed bradycardia 8 weeks after STZ injection. In order to evaluate diastolic function (Table 2), color flow guided, pulsed-wave Doppler imaging was recorded for mitral and pulmonary venous flows. We reported alterations in some diastolic parameters, including an increase in systolic-to-diastolic ratio of the pulmonary veins and duration of the atrial reversal flow. Together, these echocardiographic and electrographic findings were consistent with diabetic cardiomyopathy (Tables 1 and 2). Long-term treatment with diltiazem did not attenuate diabetes-induced cardiac dysfunction. Interestingly, end systolic volume was increased in healthy diltiazem-treated patients.
Echocardiographic Systolic Parameters in Healthy and Diabetic Rats at Baseline and 8 Weeks.a
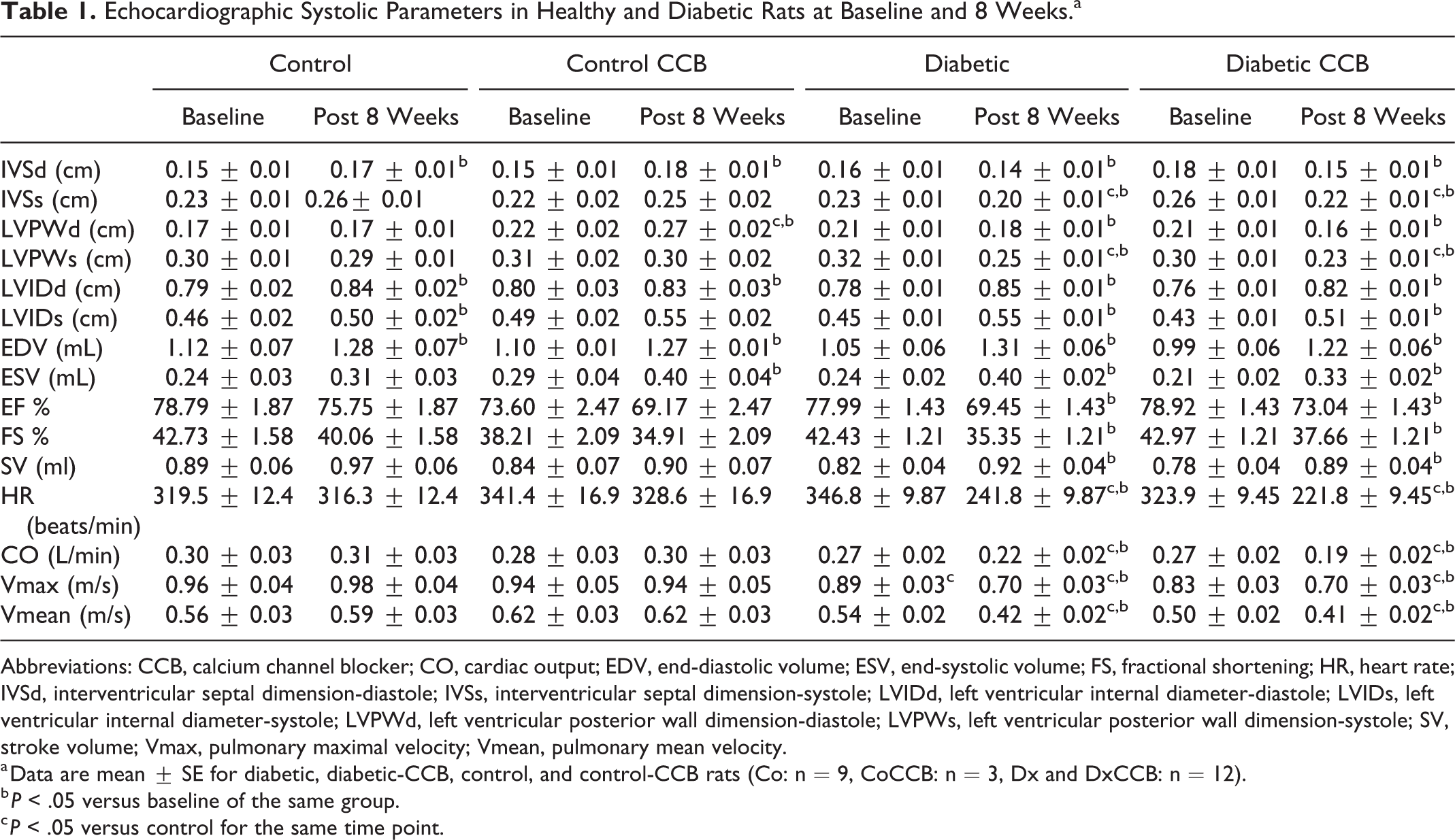
Abbreviations: CCB, calcium channel blocker; CO, cardiac output; EDV, end-diastolic volume; ESV, end-systolic volume; FS, fractional shortening; HR, heart rate; IVSd, interventricular septal dimension-diastole; IVSs, interventricular septal dimension-systole; LVIDd, left ventricular internal diameter-diastole; LVIDs, left ventricular internal diameter-systole; LVPWd, left ventricular posterior wall dimension-diastole; LVPWs, left ventricular posterior wall dimension-systole; SV, stroke volume; Vmax, pulmonary maximal velocity; Vmean, pulmonary mean velocity.
a Data are mean ± SE for diabetic, diabetic-CCB, control, and control-CCB rats (Co: n = 9, CoCCB: n = 3, Dx and DxCCB: n = 12).
b P < .05 versus baseline of the same group.
c P < .05 versus control for the same time point.
Echocardiographic Diastolic Function Parameters in Healthy and Diabetic Rats at Baseline and 8 Weeks.a

Abbreviations: CCB, calcium channel blocker; MV E/A, maximum velocity of the E wave; PVA, atrial reversal flow; S/D, systolic-to-diastolic duration ratio of the pulmonary veins.
a Data are mean ± SE for diabetic, diabetic-CCB, control, and control-CCB rats (Co: n = 9, CoCCB: n = 3, Dx and DxCCB: n = 12).
b P < .05 versus control for the same time point.
c P < .05 versus baseline of the same group.
Effect of Diltiazem Treatment on the Regulation of Glucose Transport in Striated Muscles
As we have previously shown a regionally dependent regulation of glucose transport in the heart, 37,38 we further evaluated GLUT content in the LV and right ventricle. Protein content of cardiac GLUT4, the major insulin-sensitive GLUT isoform, was decreased by 50% in the right ventricle of the untreated diabetic group compared to the age-matched control group (P = .015; Figure 2A), and a similar downward trend was observed in the LV (decrease by 45%, P = .095, Dx vs Co). While there were no significant differences between untreated and treated control groups, diltiazem treatment further worsened the diabetes-induced decrease in GLUT4 protein content of the LV (Dx-CCB vs Co: decrease by 61%, P = .024; Dx-CCB vs Dx: decrease by 28%, P = .0509). There was also significant decrease in GLUT4 protein content between treated healthy versus treated diabetic animals, in the both the LV and right ventricle (P = .024 and P = .045, respectively). In parallel, GLUT4 content was decreased by 78% in skeletal muscle of untreated diabetic versus healthy rodents (P = .049). Diltiazem treatment also further worsened the diabetes-induced decrease in skeletal muscle GLUT4 protein content (decrease by 92%, P = .052; Figure 2B). A significant correlation was observed between GLUT4 protein content in striated muscle and blood glucose concentration of (treated and untreated) diabetic and control groups (cardiac: R2 = 0.5164, P = .014; skeletal muscle: R2 = 0.3288, P = .013; Figure 2C and D), confirming that GLUT4 in striated muscle is a strong regulator of whole body glucose homeostasis.
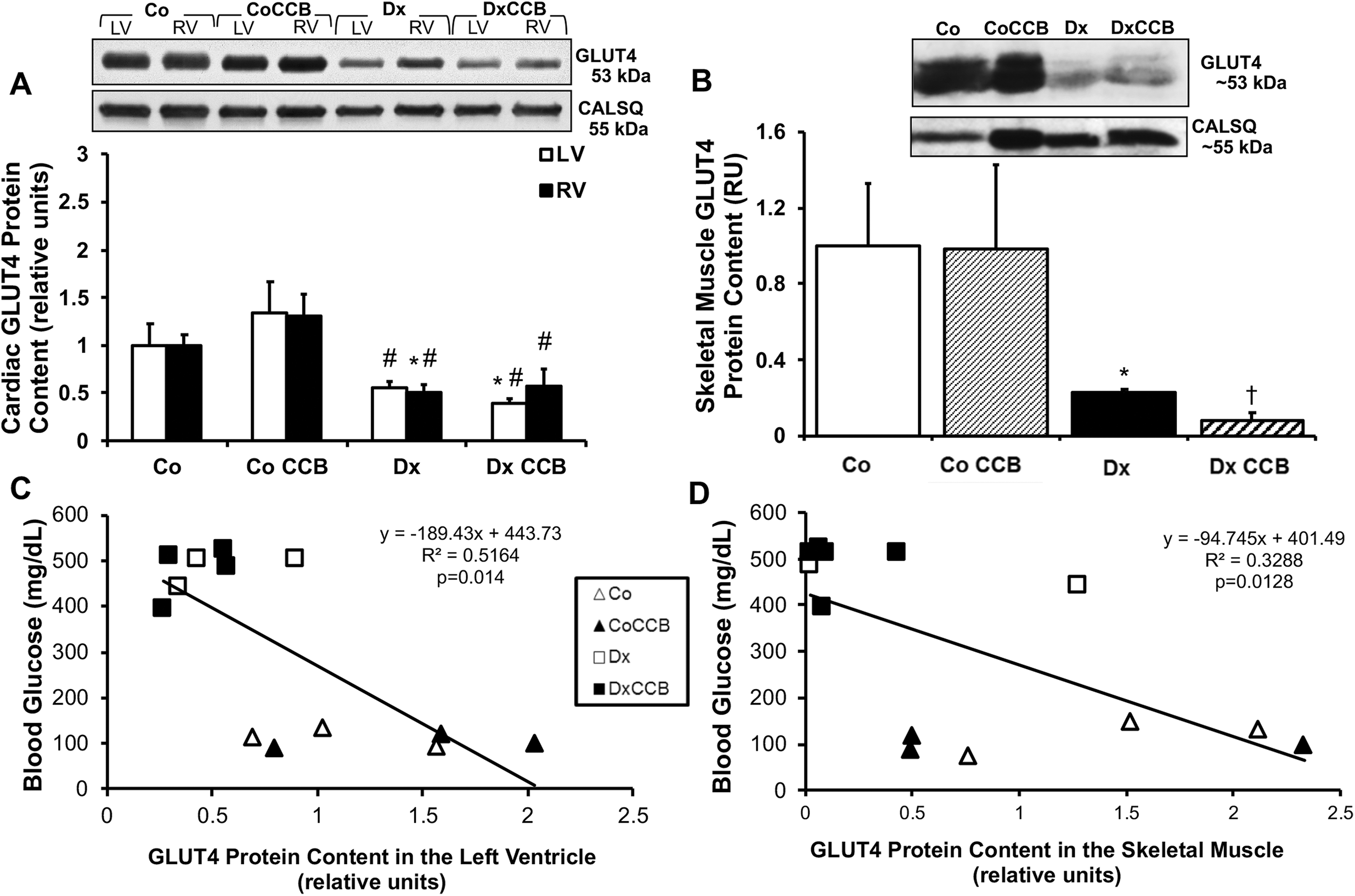
Diabetes decreases GLUT4 protein content in membrane-enriched preparation of striated muscle from diabetic rats, with a more pronounced effect after diltiazem treatment. Top panels: Representative Western blot of striated muscle from untreated healthy (Co) and diabetic (Dx), and healthy diltiazem-treated (Co-CCB) and diabetic diltiazem-treated (Dx-CCB) rats. Bottom panels: Mean ± SE total protein content in a membrane-enriched fraction of (A) GLUT4 in cardiac muscle (B) GLUT4 in skeletal muscle; values normalized to basal. Venous blood [glucose] was significantly negatively correlated with both (C) cardiac (P = .014, R2 = 0.5164), and (D) skeletal muscle (P = .0128, R2 = 0.3288) GLUT4 content. Co and CoCCB: n = 3; Dx and DxCCB: n = 3-6/group. *P < .05 versus control, #P < .05 versus Co-CCB. †P < .05 versus Dx. GLUT4 indicates glucose transporter 4; LV, left ventricle; RV, right ventricle; SE, standard error.
Since the most downstream protein regulating GLUT4 trafficking is AS160, expression of the active and total form of this protein was measured by Western blotting. There was a significant downregulation of AS160 phosphorylated protein in the heart of the treated diabetic group as compared to the treated healthy group (decrease by 61%, P = .009; Dx-CCB vs Co-CCB, Figure 3A). This protein content ratio of phosphorylated AS160 in the heart significantly correlated with blood glucose concentration (y = −191.04x + 508.95, P < .001), suggesting that the phosphorylation of AS160 is a crucial step in the regulation of glucose uptake into cardiac cell from the blood stream (Figure 3C). Similar to the results reported in the heart, diltiazem treatment significantly worsened diabetes-induced alteration in AS160 activation in the skeletal muscle, as there was a significant downregulation of AS160 phosphorylated protein in the treated diabetic rodents (decrease by 57%, P = .022; Dx-CCB vs Co, Figure 3B).

Decreased activation of AS160 in striated muscle was exacerbated by diltiazem treatment. A, B, Top panel: Representative Western blots of total and phosphorylated AS160 protein expression from a striated muscle total tissue preparation. Bottom panel: Mean ± SE of phosphorylated/total-AS160 cardiac protein content in untreated healthy (Co) and diabetic (Dx), and healthy diltiazem-treated (Co-CCB) and diabetic diltiazem-treated (Dx-CCB) rats. (A) AS160 protein content in cardiac muscle; values normalized to basal; (B) mean ± SE total protein content of total and phosphorylated AS160 protein content in skeletal muscle (n = 3-4/group). *P < .05 versus control. Values normalized to basal. C, D, Scatterplot and linear regression analysis of phospho/total AS160 content (independent variable) and venous blood [glucose] (dependent variable). (C) Correlation in cardiac muscle, showing a significant negative correlation (P < .001, R2 = 0.687; n = 3-4/group); (D) no significant correlation in skeletal muscle. SE indicates standard error; AS160, Akt Substrate of 160 kDa.
Effect of Diltiazem Treatment on Proteins Regulating Sarcoplasmic Reticulum Calcium Transport
Since the mechanisms of action of diltiazem include the inhibition of the L-type calcium channels, we investigated whether diltiazem will affect key proteins involved in excitation–contraction coupling in the diabetic myocardium. There was a significant downregulation of the protein content of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2a) pump in the heart of diabetic animals (Figure 4A). There was no significant change in SERCA protein expression between untreated and treated diabetic groups. In addition, all groups demonstrated a similar protein expression of the Ryanodine receptor in the heart (Figure 4B).

No change in the expression of major proteins regulating sarcoplasmic reticulum calcium handling in diltiazem-treated diabetic rats. Top panels: Representative Western blot of cardiac muscle from untreated healthy (Co) and diabetic (Dx), and healthy diltiazem-treated (Co-CCB) and diabetic diltiazem-treated (Dx-CCB) rats. Bottom panels: Mean ± SE total protein content in a membrane-enriched fraction of (A) SERCA2a in cardiac muscle; (B) RYR2 in cardiac muscle. Co and CoCCB: n = 3/group; Dx and DxCCB: n = 6/group. *P < .05 versus control, #P < .05 versus Co-CCB. SE indicates standard error.
Discussion
The therapeutic effects of CCBs in patients with diabetes are both fairly wide ranging and controversial. 25,26,39 -44 The current study demonstrated that administration of diltiazem at therapeutic doses to diabetic rats resulted in impairments of the regulation of glucose transport in striated muscle, which may exacerbate hyperglycemia. At the molecular level, diltiazem treatment decreased GLUT4 and AS160 protein content in striated muscle. Additionally, our animal model displayed diabetic cardiomyopathy, as evidenced by alterations in systolic and diastolic function, which was not rescued by long-term treatment with diltiazem.
Due to the blockade of L-type calcium channels, which are crucial for both the proper functioning of insulin-mediated glucose uptake and the pancreatic release of insulin, 45,46 CCBs have been found to be associated with hyperglycemia in diabetic and nondiabetic patients. 28,42 Indeed, several case studies reported CCBs to have a directly diabetogenic effect. 47,48 Although the heart is one of the main organs to utilize glucose as an energy substrate, the effect of CCB treatment on glucose transport in the heart remains to be elucidated. This is a crucial aspect to investigate when considering the clinical treatment of diabetic cardiomyopathy and arrhythmias, which are serious and common complication of diabetes. 49 Thus, we sought to examine how a common nondihydropyridine CCB, diltiazem, affected not only blood glucose levels and cardiac function in insulin-deficient rodents but also key cellular glucose transport mechanisms such as GLUT4 and AS160 in both cardiac and skeletal muscle. Notably, diltiazem is able to decrease arterial pressure without producing the reflexive cardiac stimulation caused by the other dihydropyridine class of CCBs, making it an optimal choice for this investigation. 50 However, reports on the effect of diltiazem treatment on cardiac function have been controversial with studies reporting no effect, 10 while others reporting a positive effect in some types of cardiomyopathies. 11,12 These controversial findings on the potential beneficial effects on cardiac function have also been reported for other CCBs. 51 Thus, while diltiazem did not improve cardiac function in this study, the potential detrimental effects of CCB therapy could stem from the negative consequences regarding glucose homeostasis.
Although the majority of patients with diabetes suffer from type 2 diabetes, 52 we here chose to use a type 1 diabetic animal model. It is clear that hyperglycemia alone, as in a type 1 diabetic model, is sufficient to induce severe diabetic cardiomyopathy 53 and induce significant alterations in cardiac function and glucose transport. 3,38 Additionally, it is important to identify the complications stemming from and affecting the hyperglycemic state, versus the complications caused by mild hyperglycemia, hyperinsulinemia, insulin resistance, and obesity, as in a type 2 model. Thus, a type 1 model was optimal for our investigation into the effects of a CCB on glucose toxicity and diabetic cardiomyopathy. While a future study regarding diltiazem treatment with a type 2 diabetic animal model will provide further insight, we are here able to separate other confounding variables (eg, obesity) from hyperglycemia and the ensuing diabetic cardiomyopathy.
We used an insulin-deficient animal model, achieved by injecting healthy rats with STZ, a compound that destroys the pancreatic β cells (which produce insulin). 3,5,38 Since a close relationship between the STZ dose and the severity of diabetes exists, we used a low dose of STZ to induce a mild form of diabetes and to mimic the early metabolic and cardiac events that occur in patients with diabetes, as previously described by our group. 5 In this model, we did not report the presence of cardiac fibrosis. 5 Our model was confirmed by the observed hyperglycemia and decreased body weight in the diabetic groups, which are hallmark features of insulin-deficient type 1 diabetes. Diltiazem treatment did not improve either of these markers in the treated diabetic rodents over their untreated diabetic counterparts. In fact, as is evident at the 3-day after STZ injection, the treated diabetic rodents became more hyperglycemic than the untreated rodents. This is in agreement with recent clinical trials reporting that 7% (569 out of 8098) of patients with coronary artery disease being treated with a CCB developed type 2 diabetes. 54 In addition, CCB overdosage (in particular, verapamil and diltiazem) has been associated with hyperglycemia. 28,42 Given the apparent worsened condition in the short term, one could speculate that the alteration in glucose transport in striated muscle would have been more deleterious in the short term in the diltiazem-treated diabetic group. In addition, CCB treatment worsened whole body glucose homeostasis in diabetic rodents, which may have long-term negative effects on the heart due to exacerbated glucose toxicity. Regardless of concern related to the short- versus long-term effects, one should note that we did not find any adverse effects of diltiazem on glucose transport in the control group, confirming that diltiazem treatment worsens metabolic alterations and thus may be contraindicated in patients with diabetes.
Similar to previous studies by our laboratory and others, 3,5,49 we reported that the untreated diabetic rodents displayed bradycardia, ventricular dilation, and systolic and diastolic dysfunction, the hallmark of diabetic cardiomyopathy. This common cardiac complication of diabetes often results in heart failure, which is to blame for nearly two-thirds of the deaths in the type 1 diabetic population. 55 Furthermore, the prediabetic state alone has been shown to be an independent risk factor for cardiovascular disease. 56 Thus, it is likely that prescription of CCBs to prediabetic patients could be a frequent occurrence and may also be contraindicated in this population. 57 In our study, treated diabetic animals did not display significant cardiac improvement compared to the untreated animals. At the molecular level, SERCA protein expression was decreased in the myocardium of diabetic rodents, as previously reported, 3,5 with no significant improvement following diltiazem treatment. In brief, our data indicated not only that diltiazem treatment did not improve cardiac function, but it further worsened whole body glucose dysregulation in diabetic rodents.
GLUT4 is the key player in insulin-mediated glucose transport and its protein content is decreased in insulin-sensitive tissues of patients with diabetes. 38,58 -60 Accordingly, in the current study, STZ-treated rodents had significantly less GLUT4 protein content in a crude membrane extract of striated muscle than their control counterparts. Not only was this decrease not rescued by treatment with diltiazem, but it was further worsened in both cardiac and skeletal muscle. In agreement with our findings, previous studies have shown that high-dose CCBs inhibit glucose transport in healthy skeletal muscle, 18,19,21 suggesting that reducing Ca2+ influx may attenuate glucose transport by altering Ca2+-dependent signaling pathways. Alternatively, several authors have suggested a direct inhibition of GLUTs by CCBs. 18,21,22 Previous studies investigated the effect of CCBs at a therapeutic dose on striated muscle glucose transport and found that while verapamil inhibits insulin-stimulated glucose transport in vitro in rat skeletal muscle, diltiazem had no effect. 20,61 Taken together, these data suggest that (1) calcium is a key player in the regulation of glucose uptake in striated muscle and (2) reducing Ca2+ influx, by direct inhibition of the L-type Ca2+ channels, decreases glucose transport in striated muscle through calcium-dependent signaling pathways. 16,62
The insulin- and calcium-dependent glucose transport pathways culminate in the phosphorylation of AS160, which precedes the translocation of GLUT4 to the cell surface. 38 The role of AS160 in the regulation of GLUT translocation during diabetes and insulin resistance in striated muscle is somewhat controversial. 38,63,64 Some groups have previously reported no difference of AKT and AS160 phosphorylation during diabetes in skeletal muscle. 64 Similarly, we reported no significant change in AS160 in skeletal muscle of untreated diabetic rats. In contrast, diltiazem treatment significantly decreased AS160 phosphorylation in the skeletal muscle of the treated diabetic rats. Additionally, in this study, as in others, 38,63 we reported a trend toward a decrease in AS160 activation in the heart of untreated diabetic versus control rodents. Similar to findings in skeletal muscle, we reported that diltiazem further worsened alterations in cardiac AS160 activation in the treated diabetic group. The decreased activation of AS160 paralleled the decrease in GLUT4 protein expression in striated muscle. We further reported a significant negative correlation between phospho/total AS160 protein content in the heart and blood glucose concentration, suggesting a key role for AS160 in regulating glucose transport and utilization in this highly metabolic organ. Taken together, these data suggest that an adverse effect on glycemic control is occurring in those diabetic rodents treated with diltiazem.
Conclusion
In conclusion, using an STZ-induced diabetic rat model, our data suggested that diltiazem at therapeutic doses altered key regulators of glucose transport (ie, GLUT protein expression and AS160 activation) in both the heart and skeletal muscle of diabetic rodents, which may further worsen the hyperglycemia observed in treated diabetic rodents. Diltiazem, a negative chronotropic and inotropic agent, did not rescue systolic dysfunction or bradycardia in diabetic animals. Thus, despite the wide prescription of CCBs for cardiovascular diseases, our data suggest that administering diltiazem to patients with diabetes may be contraindicated and further investigations into the appropriate administration of CCBs may be warranted.
Footnotes
Acknowledgements
We would like to thank Dr. Zahra Maria and Summer Hayes for their excellent technical assistance.
Author Contribution
MP contributed to the study execution, data collection and analysis, and preparation of the manuscript. AC and AW contributed to data collection and analysis, and preparation of the manuscript. VAL contributed to the study design and execution, data interpretation and preparation of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The article was supported by the American Heart Association, Great River affiliate (0855497D, VAL), and the Ohio State undergraduate research scholarship (MP).