
Abstract
Purpose:
Methotrexate is an anti-inflammatory drug that has been shown to have anti-ischemic effects. Our aim was to evaluate if methotrexate could reduce infarct size in patients with ST-segment elevation myocardial infarction (STEMI).
Methods:
We randomly assigned patients with STEMI to receive either methotrexate or placebo. Primary outcome was infarct size determined by calculating the area under the curve (AUC) for creatine kinase (CK) release. Secondary outcomes were AUC of CK MB (CK-MB) and AUC of troponin I; peak CK, peak CK-MB, and troponin I; B-type natriuretic peptide (BNP) level, high-sensitivity C-reactive protein (hsCRP) result, and erythrocyte sedimentation rate (ESR); left ventricular ejection fraction (LVEF); thrombolysis in myocardial infarction (TIMI) frame count; Killip score; mortality and reinfarction incidence; and incidence of adverse reactions.
Results:
We included 84 patients. Median AUC of CK was 78 861.0 in the methotrexate group and 68 088.0 in the placebo group (P = .10). Patients given methotrexate and placebo exhibited, respectively, median AUC for CK-MB of 9803.4 and 8037.0 (P = .42); median AUC for troponin of 3691.1 and 2132.6 (P = .09); peak CK of 2806.0 and 2147.0 (P = .05); peak CK-MB of 516.0 and 462.3 (P = .25); and peak troponin of 121.0 and 85.1 (P = .06). At 3 months, LVEF was lower in patients who received methotrexate (49.0% ± 14.1%) than in patients given placebo (56.4% ± 10.0%; P = .01). There were no differences in hsCRP, ESR, BNP, Killip scores, TIMI frame count, reinfarction, and mortality rates. There was a higher median serum glutamic–pyruvic transaminase levels in the methotrexate group.
Conclusion:
Methotrexate did not reduce infarction size and worsened LVEF at 3 months (Clinicaltrials.gov identifier NCT01741558).
Introduction
During myocardial ischemia, there is clear evidence of inflammatory activation, with increased proinflammatory cytokine levels. 1 Methotrexate is an anti-inflammatory drug that acts by promoting increased adenosine levels and also increases the vasodilator effect provoked by adenosine. 2 –6 Adenosine, in turn, can act in different adenosine receptors, with consequent reduction in hibernating and stunned myocardial areas, reduction in ischemia areas, reduction in myonecrosis during percutaneous coronary intervention (PCI), reduction in oxidative stress, and reduction in platelet adhesion. 7 –15 Methotrexate, on the other hand, inhibits atherogenesis in rabbits and reduced size of the infarction induced experimentally in dogs by reducing reperfusion injury. 15,16 In humans, methotrexate is associated with a reduction in rheumatoid arthritis. 17,18 Additionally, the Effects of METhotrexate therapy on the physical capacity of patients with ISchemic heart failure (METIS) trial showed that in humans there is a nonsignificant trend to improvement in functional class among patients with heart failure of ischemic etiology who are given methotrexate. 19
Although the use of anti-inflammatory agents would seem logical in ischemic heart disease, nonsteroidal anti-inflammatory drugs (NSAIDs) increase the incidence of coronary events, and the only classes of drugs with anti-inflammatory effects that are widely used in these situations are statins and low-dose aspirin. 1,20 However, recent clinical trials show that other anti-inflammatory and immunosuppressive drugs such as cyclosporine may reduce the infarct size. 21
As such, methotrexate emerges as a possible treatment option for acute myocardial infarction. Our study is designed to evaluate whether methotrexate combined with the standard treatment could reduce infarct size in patients with ST-segment elevation myocardial infarction (STEMI) undergoing primary angioplasty. We also intend to evaluate whether methotrexate could reduce inflammation in acute myocardial infarction, improve coronary flow in the culprit artery, and reduce heart failure incidence (reducing the area of ischemia, the degree of ventricular dilatation, and the extent of areas of stunned myocardium).
Materials and Methods
MethotrexaTE THerapy in ST-Segment Elevation MYocardial InfarctionS (TETHYS) is a prospective, randomized, phase 2, double-blind, placebo-controlled trial (Clinicaltrials.gov identifier NCT01741558). We conducted the trial at the emergency department of the Institute of Cardiology in Santa Catarina, Brazil. The study design has been described previously. 22 The informed consent was obtained from each patient; the study protocol conforms to the ethical guidelines of the Declaration of Helsinki, and the local ethics committee approved the trial protocol. An independent data and safety monitoring board closely monitored the trial and was responsible for conducting an interim analysis when 50% of the planned number of patients had been randomized. All drafts of the article were written by the first author and reviewed by all of the other authors.
Patients were eligible if they were 18 years or older, with onset of suggestive chest pain symptoms during the previous 12 hours, with an electrocardiogram with ST-segment elevation ≥0.2 mV in at least 2 contiguous leads and intention to treat with primary PCI. Exclusion criteria included previous myocardial infarction, heart failure, PCI in the previous 3 months, infarction with cardiac arrest or cardiogenic shock, previous renal failure (serum creatinine >2.0 mg/dL), alcohol consumption (consumption ≥20 doses per week), 23 illicit drug use, rheumatoid arthritis, neoplasia, infectious diseases, prior anemia (hematocrit below 30%), anti-inflammatories during the previous week (whether NSAIDs or other), excessive xanthine consumption (more than two and a half cups of coffee or two and a half gourds of mate), 24,25 and pregnancy. All patients provided informed consent in writing before enrollment.
Patients were randomly assigned to receive methotrexate or placebo (riboflavin sodium phosphate 0.1%) at a ratio of 1:1. Treatment and control groups were blinded, and we used riboflavin for the placebo group in order to preserve double-blinding, since methotrexate has a yellow color and riboflavin has the same color in this concentration. The randomization scheme was generated using specific software by an independent researcher who retained the code for any future need. The randomization scheme used was block randomization, with block sizes of 4 patients. Allocations were concealed using identical randomly numbered ampoules, labeled by the independent researcher with their respective sequential code numbers as generated by the software.
Patients in the methotrexate group were administered a methotrexate bolus of 0.05 mg/kg body weight immediately prior to PCI, followed by 0.05 mg/kg/h for 6 hours, while patients in the placebo group were administered 0.1% riboflavin sodium phosphate with the same parameters. The methotrexate dose was chosen on the basis of experimental data. 15 Patients in both the groups were given a 5-mg single dose of folic acid and should also have received all care set out in the guidelines. 26 After randomization and after initiating the infusion, patients were treated with PCI of the culprit vessel.
Patients were followed up while still in hospital and were seen again at 3 months to determine adverse events and clinical end points.
The primary efficacy study end point was infarct size as measured by creatine kinase (CK) area under the curve (AUC), calculated by the linear trapezoidal method from data recorded every 6 hours for 72 hours. All samples were analyzed at the local laboratory. Linear interpolation was used for missing values from intermediate time points.
Secondary efficacy end points were: AUC of troponin I and AUC of CK-MB release for 72 hours, peak CK, peak CK-MB and peak troponin I, high-sensitivity C-reactive protein (hsCRP), and B-type natriuretic peptide (BNP) at 72 hours and 3 months, 27 –29 thrombolysis in myocardial infarction (TIMI) frame count for the culprit artery, 30 erythrocyte sedimentation rate (ESR) at 72 hours, Killip score at 72 hours, left ventricular ejection fraction (LVEF) with transthoracic echocardiography made by the same echocardiographer (Vivid E9 system, General Electric, Horten, Norway) at 72 hours and after 3 months, death at 3 months, and reinfarction rate at 3 months. Safety secondary end points were changes in levels of hematocrit, hemoglobin, leukocytes, platelets, plasma creatinine, serum glutamic oxaloacetic transaminase (SGOT), serum glutamic-pyruvic transaminase (SGPT), and prothrombin time/international normalized ratio at 72 hours and incidence of side effects (gastrointestinal effects, skin changes, or pulmonary effects). 31
We determined that the enrollment of 80 patients would provide power of 80% to detect an absolute between-group difference of 109 877.0 arbitrary units in the AUC of CK release, at a 2-sided α level of .05 for the Wilcoxon rank-sum test, based on previous data and estimating a 10% loss. 21 We performed efficacy analyses on results for the modified intention-to-treat population, defined as all the patients who underwent randomization and had STEMI confirmed. We performed safety analyses on data from all patients who underwent randomization and received the study drug. We expressed quantitative variables with normal distribution as means (±SD) and used t tests to compare them. We expressed quantitative variables with nonnormal distribution as medians with interquartile ranges (IQRs) and used the Wilcoxon rank-sum test to compare them. We used Fisher exact test to compare qualitative variables as appropriate. We used a post hoc analysis of covariance (ANCOVA) and ANCOVA with rank transformation (for nonnormal variables) to detect possible interactions between treatment groups, time until PCI (total ischemic time), and for anterior wall involvement. All analyses were performed using SPSS version 13.0 for Windows, and a 2-sided significance level of .05 was specified.
Results
From April 15, 2013, to June 19, 2014, we evaluated a total of 401 patients with myocardial infarction and enrolled 84 patients (43 to receive methotrexate and 41 to receive placebo) (Figure 1). Two patients in the methotrexate group and 1 patient in the placebo group did not have STEMI, and we excluded them from the efficacy analysis before unblinding to avoid bias. There were no losses during follow-up. Baseline characteristics are summarized in Table 1 and were balanced in the 2 study groups, except for nonsignificant differences in anterior wall involvement, total ischemia time, and prevalence of hypertension.

Flow Diagram.
Baseline Characteristics of the Patients.a
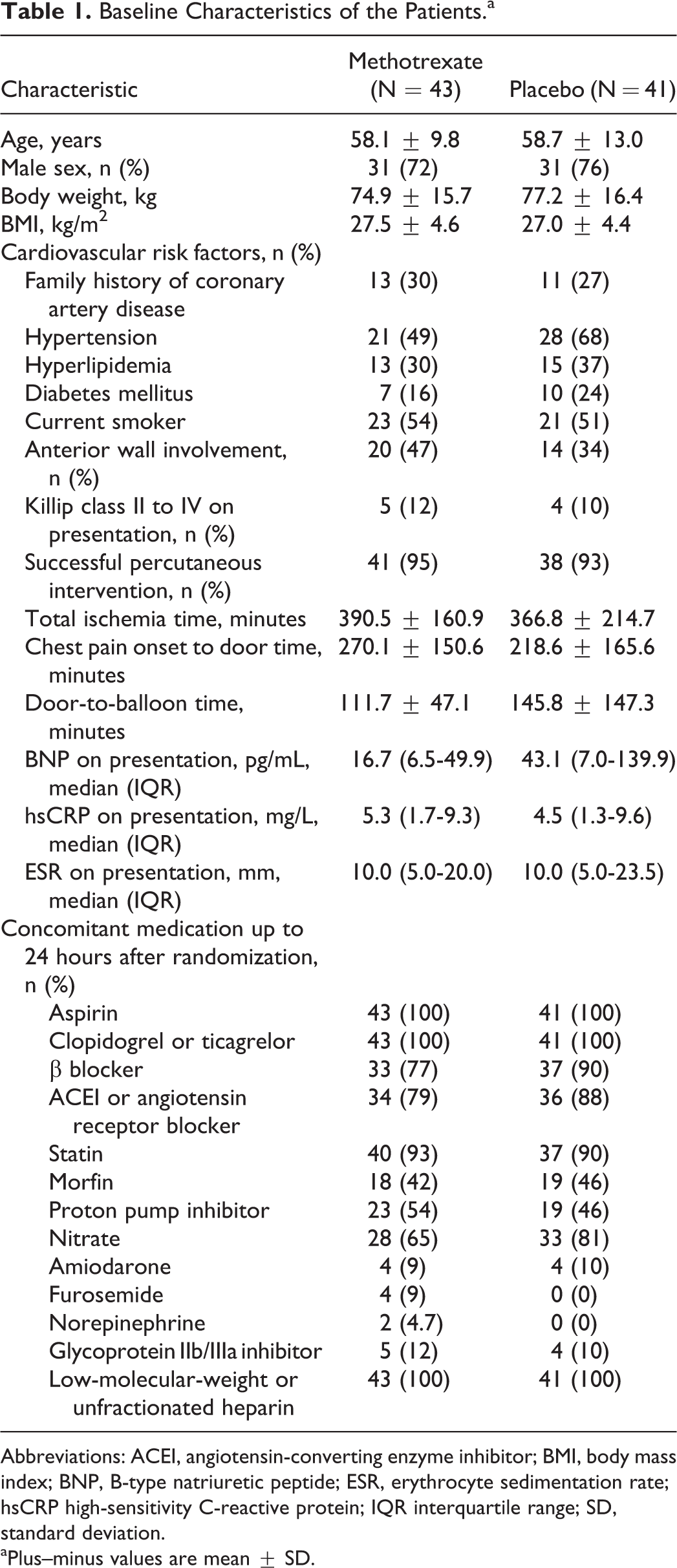
Abbreviations: ACEI, angiotensin-converting enzyme inhibitor; BMI, body mass index; BNP, B-type natriuretic peptide; ESR, erythrocyte sedimentation rate; hsCRP high-sensitivity C-reactive protein; IQR interquartile range; SD, standard deviation.
aPlus–minus values are mean ± SD.
There was a trend to a larger AUC of CK release in the methotrexate group, with a median of 78 861.0 arbitrary units (IQR: 46 633.5-124 867.5) versus 68 088.0 (IQR: 37 568.3 to 97 626.0) in the placebo group (P = .10) and a trend to a larger AUC of troponin I release, also in the methotrexate group, with a median of 3691.1 arbitrary units (IQR: 1745.4 to 6718.7) versus 2132.6 (IQR: 1088.5-4995.8) in the placebo group (P = .09). There were no differences between methotrexate and placebo groups for AUC of CK-MB release or for peak CK-MB, while peak CK and peak troponin I were higher in the methotrexate group but without significance. After adjustments for time until PCI and for anterior wall involvement (Figure 2; Table 2), there were no differences between the groups in AUC or peak levels. There were also no differences between the groups in hsCRP levels, BNP levels, ESR levels, and TIMI frame count of the culprit artery revascularization or in incidence of Killip scores II-IV at 72 hours (Table 2).
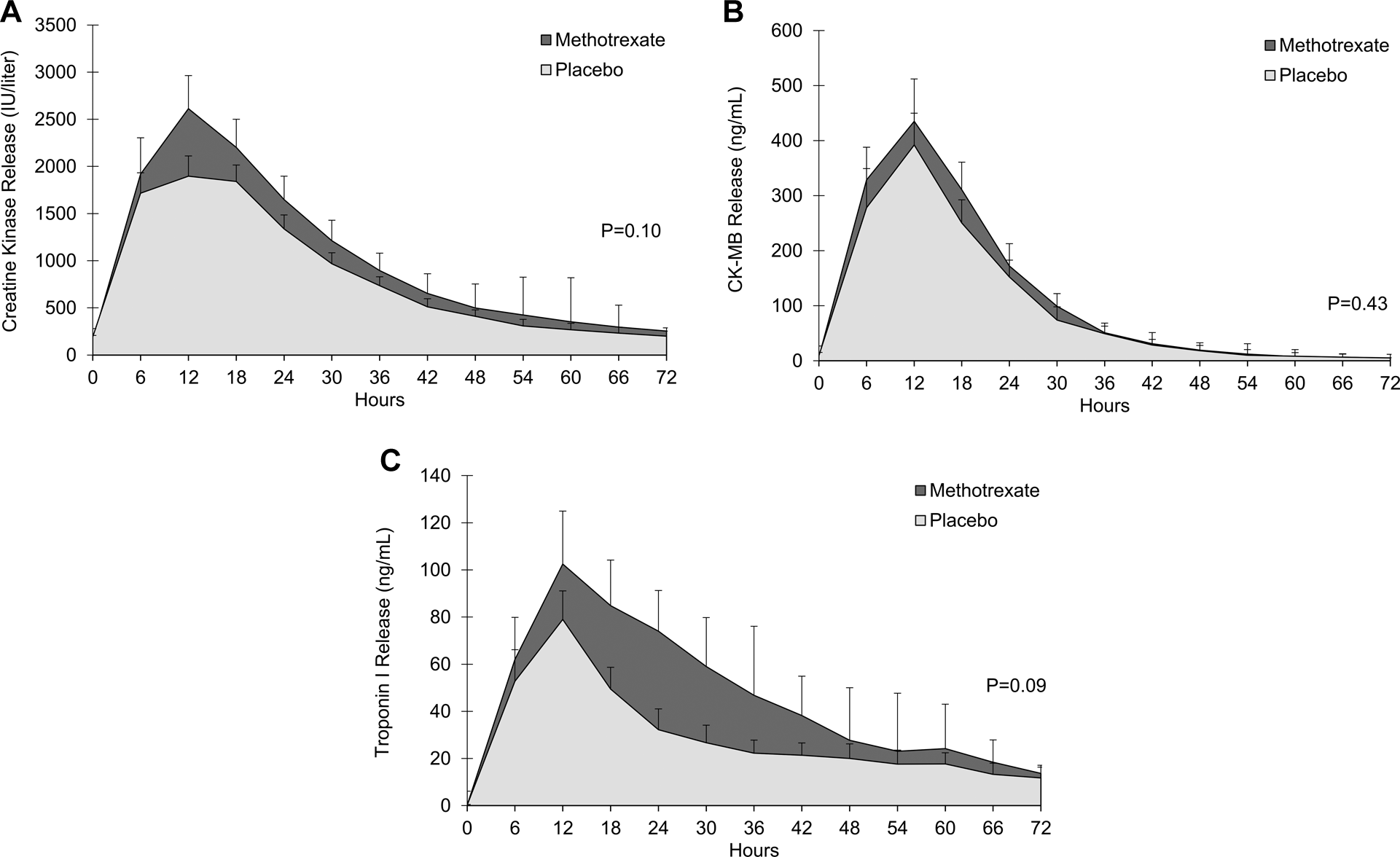
Assessment of infarct size. Serum biomarkers were measured every 6 hours up to 72 hours. Curves for methotrexate and control groups for creatine kinase (CK) release are shown in panel A, curves for CK-MB release are shown in panel B, and curves for troponin I release are shown in panel C. T bars denote standard errors.
Efficacy Outcomes.a

Abbreviations: AUC, area under the curve; BNP, B-type natriuretic peptide; CK, creatine kinase; CK-MB, creatine kinase MB; hsCRP, high-sensitivity C-reactive protein; IQR, interquartile range; LVEF, left ventricular ejection fraction; SD, standard deviation; TIMI, thrombolysis in myocardial infarction.
aPlus–minus values are mean ± SD.
bAdjusted for time until percutaneous coronary intervention (total ischemia time) and for anterior wall involvement.
c P = .04 compared with LVEF at 72 hours.
There was no difference in LVEF at 72 hours, with 49.0% ± 12.1% in the methotrexate group and 52.2% ± 11.2% for placebo (P = .21). After 3 months, however, LVEF was 49.0% ± 14.1% in patients with methotrexate treatment versus 56.4% ± 10.0% in patients on placebo (P = .01), and the lower LVEF in the methotrexate group was still significant after adjustments for time until PCI and for anterior wall involvement (Table 2). There were 4 (10%) deaths in the methotrexate group and 2 (5%) in the placebo group (P = .68). Reinfarction occurred twice (5%) in the methotrexate group and did not occur in the placebo group (P = .49).
After 72 hours, median SGPT level was higher in the methotrexate group at 57.0 (IQR: 41.0-81.0) versus 45.5 (IQR: 32.0-66.5; P = .045). There were no significant differences in levels of creatinine, hematocrit, hemoglobin, leukocytes, platelets, or SGOT. There were no differences between groups in incidence rates of other side effects (Table 3).
Safety Outcomes.a

Abbreviations: IQR, interquartile range, PT/INR, prothrombin time/international normalized ratio; SD, standard deviation; SGOT, serum glutamic oxaloacetic transaminase; SGPT, serum glutamic-pyruvic transaminase.
aAt 72 hours, except for nausea/vomiting and pulmonary infections (at 3 months); plus–minus values are mean ± SD.
Discussion
In our randomized clinical trial comparing methotrexate to placebo in patients with STEMI treated with PCI, there was a trend to greater infarction, assessed in terms of AUC of CK and AUC of troponin I and peak CK and peak troponin I, with no differences in AUC of CK-MB or peak CK-MB. There were also no differences in post-PCI coronary flow, BNP levels, or proportion of patients with Killip II or higher. Furthermore, evaluation of LVEF demonstrated no significant differences at 72 hours and greater LVEF after 3 months in the group who received placebo. These findings contrast with the experimental data showing that methotrexate promotes a reduction in experimentally induced infarction, probably in an adenosine-dependent manner. 15 The difference between our study and the experimental studies can be explained by several factors, including the facts that the experimental model is constructed by ligation of a coronary, which is quite different from the atherosclerotic process; the animals used are young and healthy, which provoke a more intense inflammatory process, and experimental models do not receive other already established anti-ischemic treatments. 32 We also cannot rule out the possibility that, unlike the experimental studies, the methotrexate doses used in our study may have been insufficient (or excessive) to achieve the correct anti-ischemic effect. 15
The difference in LVEF values that appeared by 3 months was apparently the result of greater recovery of ventricular function in the placebo group. These data, together with the nonsignificant trend to greater infarction in patients receiving methotrexate, suggest that the drug may interfere negatively with the tissue repair process, starting early on in the infarction. The reason for the possible negative effects of methotrexate remain unknown. One of the factors that could possibly have explained these findings might be the larger number of patients in the group, given methotrexate with anterior wall STEMI and who spent more time waiting for PCI. However, a post hoc analysis refuted this hypothesis, showing that the difference in LVEF remained significant even when adjusted for these variables. Methotrexate increases coronary adenosine levels, and this could bring anti-ischemic effects. Experimental data also suggest, however, the presence of adenosine-induced coronary steal through collateral circulation between nonperfused and perfused areas in the setting of coronary occlusion. Furthermore, the extent of nonperfused areas delineated by real-time myocardial contrast echocardiography increases in response to adenosine administration and could be the cause of worsening of LVEF after 3 months. 33 The complexity and redundancy of the inflammatory process in infarction could also explain the findings of our study: Blocking certain inflammatory pathways with methotrexate might be insufficient to block the entire process. This hypothesis is also supported by the fact that treatment with methotrexate provoked no reduction in ESR and hsCRP levels compared to placebo. Inflammatory process redundancy was suggested as the likely cause of failure of anti-inflammatory treatments for heart failure such as infliximab and etanercept. 34,35 The inflammatory redundancy hypothesis could also justify the failure of a range of anti-inflammatory drugs assessed in patients with unstable angina or myocardial infarction, such as methylprednisolone, 36 PG-116800 (matrix metalloproteinase inhibitor), 37 varespladip (secretory phospholipase A2 inhibitor), 38 pexelizumab (C5 complement fraction inhibitor), 39 and darapladib (phospholipase A-associated lipoprotein inhibitor). 40
Furthermore, although the whole process of inflammation in coronary artery disease and in STEMI is well known, we do not know whether inflammatory blockade is actually beneficial, especially during infarction. 1 We cannot rule out that the negative results may be consequence of study design: Patients were included up to 12 hours from symptom onset, and it is possible that, in order to expect some benefit on infarct size, treatment should be administered earlier. We also cannot rule out the possibility that the inflammatory process could be important to the early tissue repair process, since it promotes changes such as the removal of cellular debris, neovascularization, and healing in the infarcted area, and interfering with this process may be the cause of the negative results. 41,42 Experimental studies have shown, for example, that the depletion of macrophages in the myocardial infarction provokes deterioration in ventricular function and reduction in angiogenesis. 43,44 Likewise, interleukin 6 blockade in animal models increases infarction size and reduces the postinfarction ventricular ejection fraction. 45 A potential blockade of inflammatory activity in the immediate postinfarction period could explain the negative action of methotrexate in our study, although its effect on stable patients may be protective. There is experimental evidence that methotrexate may decrease the atherosclerotic process, 16,46 and evidence from observational studies of patients with rheumatic diseases shows that methotrexate reduces the incidence of infarction. 17,18,47 The potential benefit of methotrexate in stable patients with ischemia will be proven or refuted by the results of the Cardiovascular Inflammation Reduction Trial which will evaluate patients with a high prevalence of subclinical vascular inflammation (diabetes or metabolic syndrome) after a minimum period of 60 days postinfarction. 48
Although it is possible that patients who received methotrexate suffered damage to ventricular function, the drug exhibited a reasonable safety profile, with no differences in the incidence of adverse effects, except for higher median SGPT levels.
The study has some limitations. The first is the fact that we had not predicted that patients who had all of the inclusion criteria and none of the exclusion criteria might not have infarction (as in the cases of Prinzmetal angina). Despite having adopted the principle of intention to treat, a type I error could have resulted if these patients’ data had been included in the efficacy assessment, and, in view of this, the independent data and safety monitoring board suggested that these patients be excluded from the efficacy evaluation (before the unblinding process) but maintained in the safety assessment. Post hoc efficacy assessments including these patients did not change the study findings (data not shown). It is important to mention that, although the sample size was estimated based in other clinical trial, we cannot rule out that was based in a wildly optimistic reduction in infarct size. Thus, it is possible that the absence of differences in infarct size in our trial was promoted by a type II error. Our study also showed high time to reperfusion, with a mean of more than 6 hours in both the groups, which may have impacted negatively on methotrexate action: A post hoc evaluation of Acute Myocardial Infarction Study of Adenosine II (AMISTAD-II) demonstrated that adenosine (that is a product of action methotrexate) reduces mortality only when infused in reperfusion therapy within 3.17 hours. 49 The study is also limited with respect to outcomes, since imaging methods such as magnetic resonance imaging were not available, and it was only possible to evaluate infarct size using AUCs of biomarkers. This does not however invalidate the data, since these areas under curves are classically described as outcomes in other clinical trials. 21 Another important limitation was the unavailability of markers such as interleukins for analysis, limiting the possibilities for drawing conclusions regarding the effects of methotrexate on these factors. We should also point out that while our small-scale trial detected no difference in mortality or reinfarction incidence, and the study was not designed with sufficient power to demonstrate potential differences in outcomes such as these. Despite these limitations, however, our study showed careful driving and brings important contributions on the use of methotrexate in ischemic heart disease. In addition, our findings may be included in future meta-analyses on the topic.
In summary, the TETHYS trial showed that methotrexate bolus of 0.05 mg/kg followed by 0.05 mg/kg/h for 6 hours in patients with STEMI did not reduce infarct size, did not improve coronary blood flow, and did not reduce inflammation and worsened ventricular ejection fraction after 3 months. These results should discourage its use in patients with STEMI.
Footnotes
Acknowledgments
The authors thank Álvaro Steckert Filho and Kárila Scarduelli Luciano.
Author Contributions
Daniel Medeiros Moreira contributed to conception and design. Maria Emilia Lueneberg, Roberto Leo da Silva, and Tammuz Fattah contributed to conception and acquisition. Carlos Antonio contributed to conception, design, analysis, and interpretation. Daniel Medeiros Moreira contributed to acquisition, analysis, and interpretation. All authors drafted and critically revised the manuscript and gave final approval.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.