
Abstract
Cardiac remodeling caused by acute myocardial infarction (AMI) represents a major challenge for heart failure research. MiR-155 has been identified as a key mediator of cardiac inflammation and hypertrophy. In this study, we investigate the role of miR-155 in cardiac remodeling induced by AMI. We demonstrate that miR-155 expressed in cardiac fibroblasts is a potent contributor to cardiac remodeling. We reveal that in vivo, miR-155 knockout improves left ventricular function, reduces infarct size, and attenuates collagen deposition, whereas overexpression of miR-155 produces the opposite effects. MiR-155 knockout also inhibits cardiac fibroblast proliferation and differentiation into myofibroblasts. In addition, downregulation of tumor protein p53-inducible nuclear protein 1 (TP53INP1) by small interfering RNA reverses the effects of miR-155 knockout on cardiac fibroblasts. Our data reveal that knockout of miR-155 in cardiac fibroblasts improves cardiac remodeling by targeting TP53INP1, which may be a novel treatment strategy for cardiac remodeling.
Keywords
Introduction
Acute myocardial infarction (AMI), which is one of the most common cardiovascular diseases, is the leading cause of death due to coronary artery disease in both developed and developing countries. 1 According to the American Heart Association, approximately 15 million patients are diagnosed with myocardial infarction (MI) every year. 2 Although considerable advances in the treatment of AMI, 3 parallel increases in the morbidity and mortality induced by heart failure continue to be of concern. 4 Adverse cardiac remodeling after MI contributes to impaired ventricular function and heart failure, which are two of the main reasons for increased mortality. 5
Fibroblasts are able to differentiate into contractile myofibroblasts with smooth muscle features and play a critical role during wound healing at the infarct site. 6 The deposition of collagen secreted by myofibroblasts is essential for the structural integrity of the infarcted heart and for the prevention of left ventricular (LV) rupture 7 ; however, the overproliferation of fibroblasts and their differentiation into myofibroblasts in areas far from the infarct site lead to excessive deposition of extracellular matrix proteins, especially collagen, and increased myocardial stiffness, which results in progressive diastolic dysfunction and heart failure. 8 Thus, fibrosis is a major determinant of progressive ventricular remodeling after MI. Recently, antifibrotic strategies, such as angiotensin-converting enzyme inhibition and angiotensin-receptor antagonism, have been demonstrated to be beneficial for the modulation of cardiac remodeling by attenuating the development of progressive perivascular fibrosis 9 ; however, new, more effective strategies to prevent progressive remodeling and heart failure, such as the use of microRNAs (miRNAs), need to be explored.
MicroRNAs, a class of short (approximately 22 nt), noncoding RNAs, regulate gene expression at the posttranscriptional level by inhibiting messenger RNA (mRNA) translation. 10 Over the last several years, miRNAs have been accepted as essential intracellular mediators of normal cardiac function, and their aberrant expression is associated with cardiovascular disease. 11 Previous studies have shown that several fibroblast-enriched miRNAs, including miR-21 and miR-29, are involved in the development of fibrosis. 12-13 Antagomir-mediated silencing of miR-21 was demonstrated to prevent cardiac remodeling and failure. 14 Upregulation of miR-155 has been implicated in inflammatory diseases, including rheumatoid arthritis, 15 multiple sclerosis, 16 and heart disease. 17 Xie et al reported that pretreatment with a high dose of rosuvastatin reduced the incidence of cardiovascular events and levels of inflammatory markers in patients with acute coronary syndromes who were receiving percutaneous coronary intervention, and this effect involved the suppression of miR-155. 17 Additionally, serum levels of miR-155 were found to be approximately 4-fold higher in patients who experienced cardiac death within 1 year after discharge compared to those who did not. 18 MicroRNAs appear to be a promising candidate for a novel treatment of AMI; however, the role of miR-155 in cardiac remodeling and the mechanisms underlying its effects, especially those related to cardiac fibroblasts, remain unclear.
In this study, we investigate the role of miR-155 in cardiac remodeling after AMI. We demonstrate that miR-155 knockout (KO) inhibits the proliferation of cardiac fibroblasts and their differentiation into myofibroblasts and reduces collagen deposition. These effects involve the target of miR-155, tumor protein p53-inducible nuclear protein 1 (TP53INP1). Our data reveal that miR-155 KO in cardiac fibroblasts improves cardiac remodeling by targeting TP53INP1, which may be a novel strategy for the management of cardiac remodeling.
Materials and Methods
Animals
Male miR-155 KO (miR-155−/−), miR-155 knockin (miR-155+/+), and wild-type (WT) C57BL/6J mice (10-12 weeks old) were purchased from the Model Animal Center of Wuhan University. The miR-155−/− and miR-155+/+ mice were generated from C57BL/6J mice. The WT, miR-155−/−, and miR-155+/+ mice were randomly divided into 2 groups, the AMI group and the sham group. The number of animals in each group is shown in Table 1. The animal model of AMI was established via ligation of the left anterior descending coronary artery (LAD). The mice were intraperitoneally anesthetized with sodium pentobarbital (0.05 mg/g). Under sterile conditions, the heart was exposed through thoracotomy, and then the pericardium was gently removed. Acute MI was then induced by ligating the midportion of the LAD with 8-0 Prolene suture (Johnson & Johnson, New Brunswick, New Jersey). The sham group underwent the same surgical procedure without ligation of the LAD. After ligation of the LAD for 1, 7, or 14 days, fractional shortening (FS), left ventricular end-systolic diameter (LVESD), and left ventricular end-diastolic diameter (LVEDD; in mm) were monitored by a laser Doppler perfusion monitor (Perimed AB, Stockholm, Sweden). At 14 days after AMI, the mice were euthanized, and the hearts were collected for histological, protein, and mRNA analyses. Adult mouse cardiomyocytes and fibroblasts from WT, miR-155+/+, and miR-155−/− mice were isolated and collected as described previously 19 at the indicated time point after surgical treatment.
Number of Animals Included in the Study.

Abbreviations: AMI, acute myocardial infarction; WT, wild type.
Cell Culture and Treatments
Primary cultures of neonatal mouse cardiac fibroblasts were prepared from WT C57BL/6J, miR-155+/+, and miR-155−/− mice by dissociation of neonatal (1-2 days) hearts as described previously. 19 Briefly, the hearts were directly removed from neonatal mice (1-2 days), anesthetized with ether and killed by decapitation, and then the heart tissues were digested and centrifugated, and ventricular fibroblasts were isolated from the supernatant containing the myocyte-depleted fraction and passaged twice to remove endothelial cells. The cardiac fibroblasts were cultured in Dulbecco modified Eagle medium (Life Technologies, Carlsbad, California) supplemented with 10% fetal bovine serum (Gibco, Grand Island, New York) under an atmosphere of 5% CO2 and 95% air at 37°C. The adult cardiac fibroblast and cardiomyocytes were prepared as described previously. 19 Briefly, adult mice were anesthetized with ether, and the hearts were rapidly excised. After perfusion, digestion, and centrifugation, the pellet contained the myocyte-rich fraction, and the supernatant contained the fibroblast-rich fraction. The myocytes and fibroblast were collected for total RNA preparation.
Transforming growth factor β (TGF-β) was purchased from Sigma-Aldrich (St Louis, Missouri). Transforming growth factor β (5 ng/mL) was added to the primary cardiac fibroblasts, which were then cultured for 24, 48, or 72 hours.
Ectopic expression of miR-155 was achieved by transfecting WT cardiac fibroblasts with miR-155 mimics or inhibitors (RiboBio, Guangzhou, China) through the use of Lipofectamine 2000 (Life Technologies) according to the manufacturer’s instructions.
Small interfering RNA (siRNA)-mediated downregulation of TP53INP1 expression in primary neonatal miR-155−/− mouse cardiac fibroblasts was achieved by transfecting the cells with RNA oligonucleotides using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s instructions. The negative control (NC) siRNA and the siRNA against TP53INP1 were synthesized by Sangon (Sangon, Shanghai, China). For TP53INP1 knockdown, the following 3 siRNA oligonucleotides were used: TP53INP1-1, CACUAAUAACCC GGUUGACU; TP53INP1-2, GUGCCUACUACAGAGAAGA; and TP53INP1-3, CGGUGUG AGCUGUUUACUU.
2,3,5-Triphenyl Tetrazolium Chloride (TTC) Staining
TTC staining was performed to quantify infarct size. After assessing cardiac function, the mice were anesthetized with sodium pentobarbital and killed. The ventricles were collected and sliced into 2-mm thick slices. The slices were incubated in 2% TTC (Sigma-Aldrich) for 30 minutes at 37°C. The area unstained by TTC indicated the infarct area, which was measured using Image-Pro Plus 5.0. Infarct size (%) was expressed as infarct size divided by the left ventricle area.
Masson Trichrome Staining
The hearts were fixed in 4% formaldehyde and embedded in paraffin. Then, the tissues were longitudinally sectioned into 4-μm sections. The sections were stained with Masson trichrome stain (Sigma-Aldrich) to detect collagen deposition. The collagen-enrich tissues were stained blue, the nuclei were stained black, and the background myocardium was stained red. Each staining method was performed according to the manufacturer’s instructions. The stained sections were dehydrated in a graded alcohol series, cleared in xylenes, and then coverslipped. The stained sections were photographed using an Olympus BX50 compound microscope (Olympus Optical, Tokyo, Japan).
Immunohistochemical Staining
The mice were euthanized at each of the indicated time points. The hearts were collected and fixed in 4% formalin, embedded in paraffin, and sliced into 4-µm thick sections. The slides of heart sections were deparaffinized and hydrated. A 3% hydrogen peroxide solution was used to inactivate endogenous peroxidases. The antigens were retrieved in citric acid buffer (pH 6.0) by microwave heating. The slices were blocked with normal goat serum and incubated with a primary antibody, rabbit polyclonal anti-brain natriuretic peptide (BNP, 1:150 dilution; Bioss, Beijing, China), or anti-cardiac troponin I (1:100 dilution; Abcam, Hong Kong) overnight at 4°C. In the NC experiments, the primary antibodies were replaced with phosphate-buffered saline (PBS). The slices from all of the groups were then washed with PBS and incubated with an anti-rabbit secondary antibody (Maixin, Fuzhou, China) for 2 hours at room temperature, and then, the slices were washed with PBS twice. The signals were visualized using a DAB Detection kit (Maixin). Finally, the slices were counterstained with hematoxylin.
Immunofluorescence Staining
The primary cardiac fibroblast slices were washed with mixture of Tris-buffered saline and Tween 20 (TBST) and fixed in 4% paraformaldehyde. The slices were then blocked with 5% normal goat serum and incubated with a mouse monoclonal anti–α-smooth muscle actin (α-SMA) primary antibody (1:200 dilution; Boster, Wuhan, China) overnight at 4°C. The slices were then washed with TBST and incubated with an Alexa Fluor 488-conjugated goat antimouse secondary antibody (Life Technologies) for 60 minutes at 37°C. Then, the slices were washed with TBST and counterstained with 4′,6-diamidino-2-phenylindole (1:1000 dilution; Sigma) for 5 minutes. The samples were visualized under a microscope (Nikon, Tokyo, Japan) using the program DP2-BSW.
Quantitative PCR Assay
Total RNA was extracted from the indicated tissues or cells using a Qiagen RNeasy kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Expression of TP53INP1, collagen I, collagen III, and caspase-3 mRNA was detected by quantitative PCR (qPCR) using SYBR Green/Fluorescein qPCR Master Mix (Thermo Fisher Scientific, Waltham, Massachusetts). β-Actin expression was used as an endogenous control. The specific primers were used as following: TP53INP1, F: 5′-CTCCTGTTTACCTGCATCTTT-3′, R: 5′-GGACTTGTTTCCACCTTGA TAG-3′; collagen I, F: 5′-CGCCATCAAGGTCTACTGC-3′, R: 5′-GAATCCATC GGTCATGCTCT-3′; collagen III, F: 5′-CCCACAGCCTTCTACACCT, R: 5′-ACCCATTCCTCCCACTCC-3′; caspase-3, F: 5′-TCTGACTGGAAAGCCG AAAC-3′, R: 5′-CTGGATGAACCACGACCC-3′; β-actin, F: 5′-CACGATGGA GGGGCCGGACTCATC-3′, R: 5′-TAAAGACCTCTATGCCAACACAGT-3′. The MiScript Reverse Transcription kit (Qiagen) was used to reverse transcribe RNA into complementary DNA, and the MiScript SYBR-Green PCR kit (Qiagen) was used for real-time PCR to detect the expression of miR-155. Specific primer sets for miRNA-155 and U6 were purchased from GeneCopoeia (Guangzhou, China). Expression of U6 was used as an endogenous control. The 2−ΔΔCT method was used to analyze the data.
Western Blotting
Protein was extracted from the indicated cells in radioimmunoprecipitation assay buffer, and a 60 μg sample was separated on 10% sodium dodecyl sulfate–polyacrylamide gels to quantify the levels of TP53INP1, collagen I, collagen III, cleaved caspase-3, and β-actin. β-Actin was used as an endogenous control. The proteins were transferred to polyvinylidene difluoride membranes (Millipore, Danvers, Massachusetts) and then incubated with primary antibodies (rabbit polyclonal anti-TP53INP1, 1:200 dilution and mouse monoclonal anti-collagen I and anti–caspase-3, 1:200 dilution; Santa Cruz, Dallas, Texas) overnight at 4°C. Then, the membranes were washed with TBST and incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies (Boster, Wuhan, China). The signals were visualized using enhanced chemiluminescence substrates (Thermo Fisher Scientific) and quantified using BandScan software (Glyko Biomedical Ltd, Candiac, Canada).
Dual-Luciferase Reporter Assay
Wild-type and mutant (MUT) forms of the 3′ untranslated region (3′-UTR) of TP53INP1 were inserted downstream of the dual-luciferase reporter vector, as shown in Figure 1A and B. For the luciferase assay, 5 × 104 WT cardiac fibroblasts were plated and cultured in 96-well plates to reach approximately 80% confluence. The fibroblasts were cotransfected with 50 nmol/L miR-155 mimics or miR-155 inhibitors and 25 ng of the WT/MUT 3′-UTR of TP53INP1 dual-luciferase reporter vector using Lipofectamine 2000 (Life Technologies). After 48 hours of transfection, a Dual-Luciferase Reporter Assay System (Promega, Madison, Wisconsin) was used to detect luciferase activity using a GloMax-Multi+ Luminometer (Promega). Luciferase activity was normalized to Renilla luciferase activity.

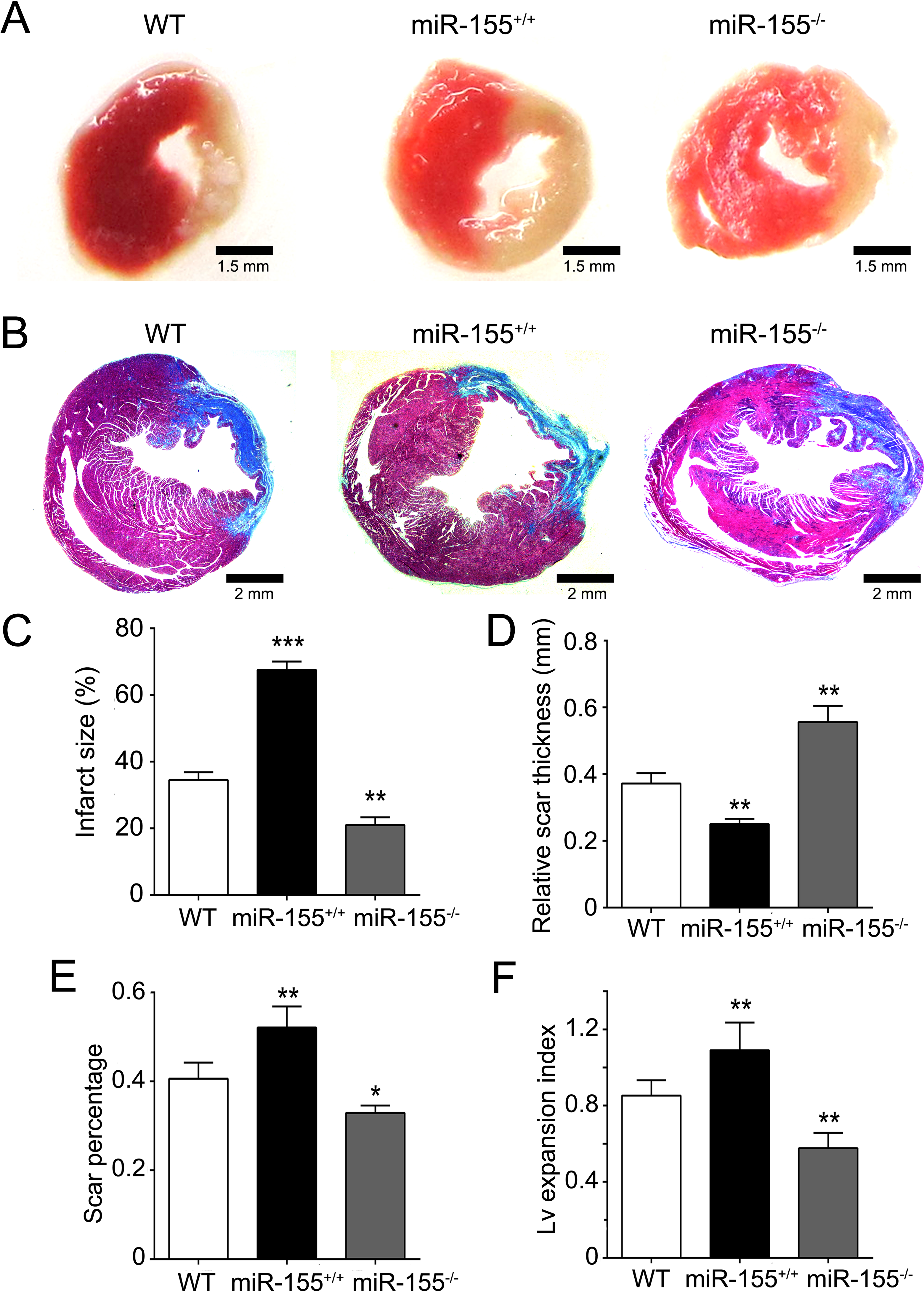
MiR-155 knockout (KO) mice are protected from left anterior descending coronary artery (LAD) ligation-induced myocardial infarct and collagen deposition. A, Representative images of trimethyl tetrazolium chloride (TTC) staining of the left ventricular (LV) infarct area at day 14 after acute myocardial infarction (AMI) induction. Infarct areas were stained in blue and healthy myocardium in red. Bar = 1.5 mm. B, Representative images of Masson trichrome-stained sections at day 14 after AMI induction. Collagen-rich areas (scar tissue) are colored in blue and healthy myocardium in red. Bar = 2 mm. C, Percentage of infarct size divided by the total LV tissue. D, Relative scar thickness (scar thickness/wall thickness). E, Scar percentage of total LV circumference calculated as scar length divided by the whole LV circumference. F, The LV expansion index ([LV cavity area/whole LV area]/relative scar thickness). The data are presented as the mean ± standard deviation (SD). *P < .05, **P < .01, and ***P < .001 versus wild type (WT).
Methylthiazol Tetrazolium Assay
The methylthiazol tetrazolium (MTT) assay was used to assess cell proliferation. After the indicated treatments, 5 × 103 cells were seeded into each well of the 96-well plates and cultured for 24, 48, and 72 hours. At the end of each time point, the cells were incubated with MTT (Sigma-Aldrich) for an additional 4 hours at 37°C. Then, 150 μL of dimethyl sulfoxide was added to each well, and the plates were incubated for 10 minutes at room temperature. The optical density was detected at 570 nm using an enzyme immunoassay analyzer (Synergy H1; BioTek, Winooski, Vermont).
Measurement of Infarct Size, Relative Scar Thickness, Scar Percentage of Whole LV Circumference, and LV Expansion Index
The infarct size, scar percentage of whole LV circumference, relative scar thickness, and infarct expansion index were quantified as described previously 20 using ImageJ software (National Institutes of Health, http://rsb.info.nih.gov/ij/links.html). For each heart, the infarct size was calculated as the mean value for the 3 analyzed sections. Thickness of scar and septum was measured at 3 different random sites, and relative scar thickness was calculated as the mean scar thickness/septum thickness. Scar percentage of whole LV circumference was calculated as scar length divided by LV circumference. The LV expansion index was calculated as the LV cavity area/(whole LV area/relative scar thickness).
Statistical Analysis
The data are expressed as mean ± standard error of the mean (SEM) and were analyzed by Student t test, one-way analysis of variance (ANOVA), or 2-way ANOVA depending on the experimental design. Statistical significance was defined for P values of <.05.
Results
MiR-155 KO Improves AMI-Impaired LV Function
To explore the role of miR-155 in AMI, we subjected WT, miR-155+/+, and miR-155−/− mice to AMI by permanently ligating the LAD. A sham operation was used as the control. As shown in Table 2, body weights were comparable across the groups before and after the AMI and sham operations; however, by days 7 and 14, LVESD and LVEDD were significantly larger and FS was significantly lower in the WT and miR-155+/+ AMI groups compared to the WT and miR-155+/+ sham groups, whereas these differences were not observed between the miR-155−/− sham and miR-155−/− AMI groups. Moreover, LVESD and LVEDD were significantly smaller and FS was dramatically higher in the miR-155−/− AMI group compared to the miR-155+/+ and WT AMI groups. These findings suggest that miR-155 KO significantly improves AMI-impaired LV function.
Echocardiography Measurements at Baseline and at 1, 7, and 14 Days After MI.

Abbreviations: AMI, acute myocardial infarction; BW, body weight; D 1, 1 day after ligation of LAD; D 7, 7 days after ligation of LAD; D 14, 14 days after ligation of LAD; FS, fractional shortening; LAD, left anterior descending coronary artery; LVEDD, left ventricular end-diastolic diameter; LVESD, left ventricular end-systolic diameter; MI, myocardial infarction; miR-155, microRNA-155; n, number of animals per group; −/−, knockout; +/+, knockin; WT, wild type.
a P < .05 versus sham.
b P < .05 versus miR-155+/+AMI.
c P < .05 versus WT AMI.
MiR-155 KO Reduces Infarct Size and LV Expansion Index and Increases Scar Thickness
To investigate whether miR-155 KO favorably attenuates myocardial remodeling, we performed TTC and Masson trichrome staining to examine the involvement of miR-155 in postinfarction cardiac remodeling. As shown in Figure 2 A and C, infarct size was significantly decreased in the miR-155−/− mice compared to the WT mice at day 14 after LAD ligation. In contrast, the infarct size of the miR-155+/+ mice was significantly increased compared to that of the WT mice. Masson trichrome staining (Figure 2B) revealed that the mice in the miR-155−/− group had a higher relative scar thickness at 14 days after MI surgery compared with mice in the WT and miR-155+/+ groups (Figure 2D). In contrast, the data demonstrated that the scar percentage of whole LV circumference and LV expansion index was significantly lower in the miR-155−/− group compared with the WT and miR-155+/+ groups, indicating a lower degree of dilation (Figure 2E and F). Moreover, we also analyzed the expression of 2 markers of myocardial injury, BNP and cardiac troponin I. We found that the expression of these 2 markers was significantly lower in the miR-155−/− AMI group than in the WT AMI and miR-155+/+ AMI groups (Figure 3), indicating that miR-155 KO reduced AMI-mediated myocardial injury. Thus, these results suggest that miR-155 KO attenuates postinfarction cardiac remodeling.
MiR-155 knockout (KO) protects against left anterior descending coronary artery (LAD) ligation-induced cardiac injury. Representative images of cardiac sections of brain natriuretic peptide (BNP) and cardiac troponin 1 (CT1) immunoreactive cells at day 14 after LAD ligation (upper). Bar = 100 μm; quantification of integrated optical density (IOD) value. The data are presented as the mean ± standard deviation (SD). *P < .05, **P < .01 versus wild type (WT).
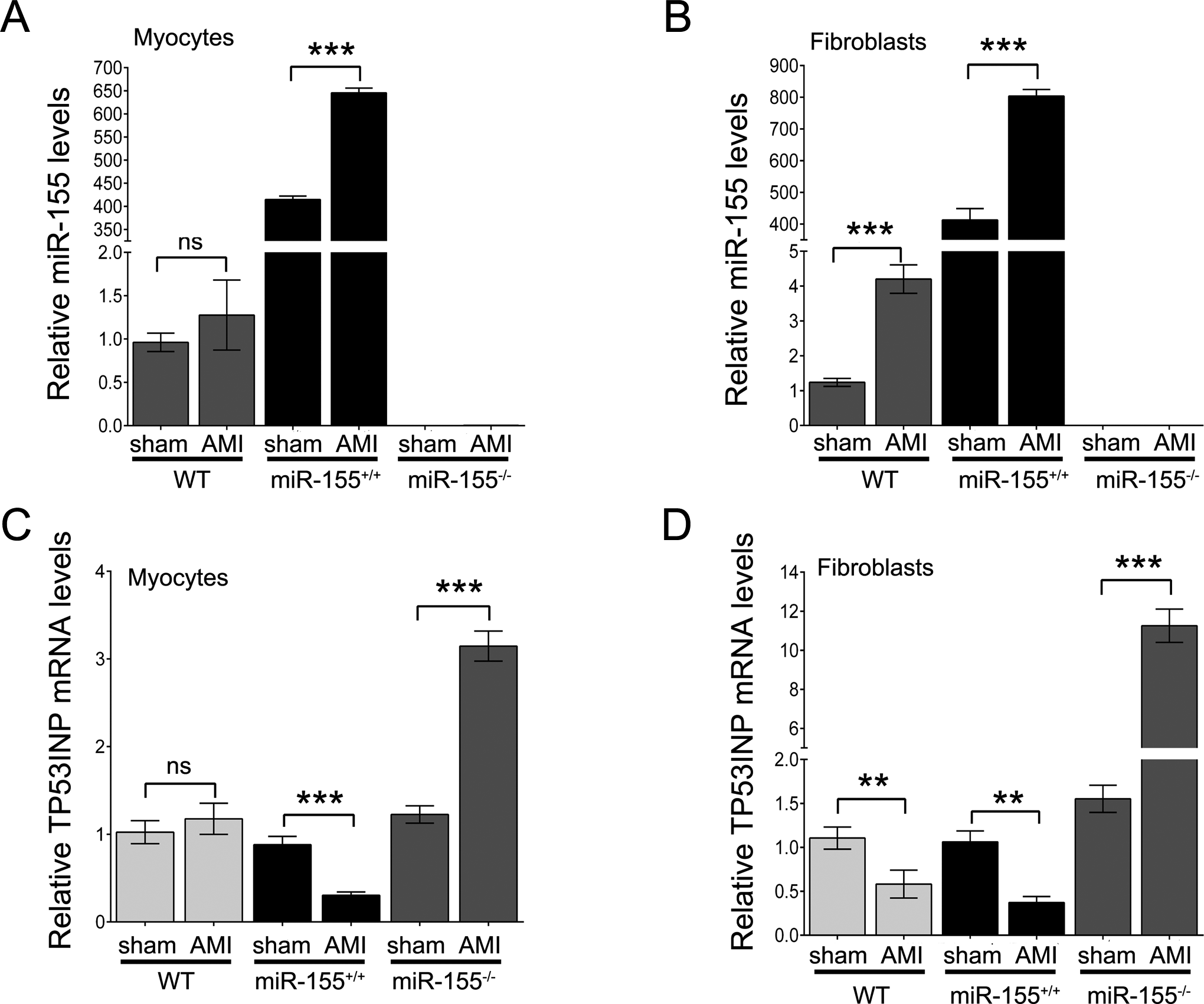
MiR-155 knockout (KO) downregulates tumor protein p53-inducible nuclear protein 1 (TP53INP1) expression in cardiac fibroblasts. A, The expression of miR-155 in cardiomyocytes at day 14 after left anterior descending coronary artery (LAD) ligation. B, The expression of miR-155 in cardiac fibroblasts at day 14 after LAD ligation. C, The messenger RNA (mRNA) expression of TP53INP1 in cardiomyocytes at day 14 after LAD ligation. D, The mRNA expression of TP53INP1 in cardiac fibroblasts at day 14 after LAD ligation. The data are presented as the mean ± standard deviation (SD). *P < .05, **P < .01, and ***P < .001; ns, no significance.
Acute MI Upregulates miR-155 Expression and Downregulates TP53INP1 Expression
Cardiac fibroblasts play an important role in cardiac remodeling, so we further investigated the cell types in which miR-155 expression contributes to cardiac remodeling. We isolated cardiac fibroblasts and cardiac myocytes from WT, miR-155+/+, and miR-155−/− mice at 14 days after sham surgery or LAD ligation. We found that AMI slightly upregulated the expression of miR-155 in the cardiac myocytes derived from the WT mice, whereas AMI significantly upregulated the expression of miR-155 in the cardiac fibroblasts and the cardiac myocytes that were isolated from the WT and miR-155+/+ mice. We did not detect the expression of miR-155 in the cardiac myocytes and fibroblasts that were isolated from the miR-155−/− mice (Figure 4A and B). Furthermore, we also analyzed the expression of TP53INP1, a putative target of miR-155. Interestingly, TP53INP1 expression in the cardiac fibroblasts was significantly downregulated following AMI compared to the sham control operation, whereas there was no significant change in cardiac myocyte TP53INP1 expression between the sham and AMI groups of WT mice. In both the cardiac myocytes and fibroblasts isolated from the miR-155+/+ mice, the expression of TP53INP1 was significantly reduced, and miR-155 KO dramatically induced TP53INP1 expression in the cardiac fibroblasts (Figure 4C and D). These results indicate that AMI significantly increases miR-155 expression and leads to the subsequent downregulation of TP53INP1 in fibroblasts of WT mice.
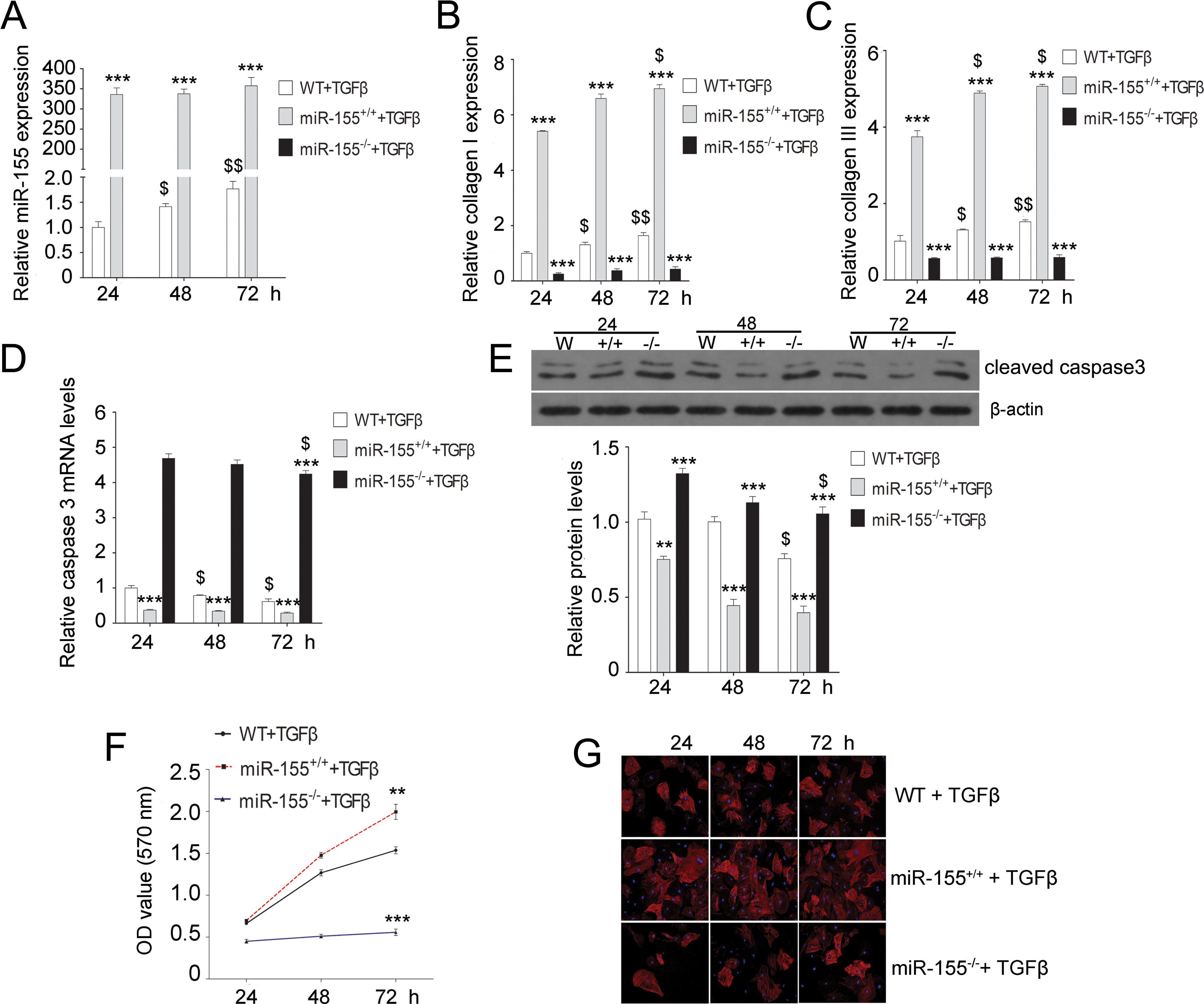
MiR-155 knockout (KO) decreases the expression of collagen and cleaved caspase-3 and inhibits cell proliferation induced by transforming growth factor β (TGF-β) in cardiac fibroblasts. The expression of miR-155 (A) and the messenger RNA (mRNA) levels of collagen I (B), collagen III (C), and caspase-3 (D) in cardiac fibroblasts at different time points after TGF-β treatment. E, Quantification of the protein expression of cleaved caspase-3 in cardiac fibroblasts at different time points after TGF-β treatment. F, Methylthiazol tetrazolium (MTT) assay-based measurements of cell proliferation in cardiac fibroblasts at different time points after TGF-β treatment. G, Immunofluorescence staining for α-smooth muscle actin (α-SMA; red) with 4′,6-diamidino-2-phenylindole (DAPI) counterstaining (blue) in mouse cardiac fibroblasts stimulated with TGF-β (magnification, ×200). The data are presented as the mean ± standard deviation (SD). *P < .05, **P < .01, and ***P < .001 versus wild type (WT) $P < .05 and $$P < .01 versus 24 hours.
MiR-155 KO Diminishes the Effects of TGF-β on Collagen I/III, Cleaved Caspase-3, and α-SMA Expression and on Cell Proliferation in Cardiac Fibroblasts
Transforming growth factor β is an activator of myocardial fibrosis, which subsequently promotes cardiac remodeling. We next investigated the role of miR-155 in myocardial fibrosis. We stimulated cardiac fibroblasts with TGF-β and found that TGF-β stimulation significantly increased the expression of miR-155 in WT cardiac fibroblasts in a time-dependent manner, and miR-155 was overexpressed in the miR-155+/+ group, but it could not be detected in the miR-155−/− group (Figure 1A). Furthermore, in the WT cardiac fibroblasts, mRNA expression of collagen I/III was dramatically upregulated with increased exposure to TGF-β. The expression was also higher in the miR-155+/+ group than in the WT group; however, miR-155 KO diminished the TGF-β–mediated upregulation (Figure 1B and C). In the WT group, the expression of caspase-3 mRNA and cleaved caspase-3 protein was dramatically and time-dependently downregulated by TGF-β. The expression was also lower in the miR-155+/+ group than in the WT group. In contrast, miR-155 KO significantly reversed the TGF-β–mediated downregulation of caspase-3 (Figure 1D and E). We then measured cardiac fibroblast proliferation after TGF-β treatment. The results revealed that overexpression of miR-155 promoted cell proliferation and that KO of miR-155 significantly inhibited cardiac fibroblast proliferation (Figure 1F). Moreover, we assessed α-SMA expression via an immunofluorescence assay. As shown in Figure 1G, TGF-β treatment increased α-SMA expression in the WT group. The expression of α-SMA was significantly higher in the miR-155+/+ group compared to the WT group, but the expression in the miR-155−/− group was significantly lower than in the miR-155+/+and WT groups.
MiR-155 Directly Targets TP53INP1
To identify whether the 3′-UTR of TP53INP1 possessed a direct target site for miR-155, a bioinformatics tool (TargetScan, www.targetscan.org) was used to predict the binding sites (Figure 5A). As shown in Figure 5A and B, WT and MUT forms of the 3′-UTR of TP53INP1 were inserted into a dual-luciferase reporter vector. Using a dual-luciferase reporter assay, we found that the luciferase activity of the WT 3′-UTR of TP53INP1 was significantly repressed in the pre–miR-155 transfectants compared to the NC group. Moreover, the miR-155-mediated repression of luciferase activity was abolished by the MUT 3′-UTR of TP53INP1 (Figure 5C). These results demonstrate that miR-155 directly targets TP53INP1.

MiR-155 directly targets tumor protein p53-inducible nuclear protein 1 (TP53INP1). A, The predicted miR-155 binding site (bold and underline) within the TP53INP1 3′ untranslated region (3′-UTR) and its mutated (MUT) version (italics and strikethrough). B, Representation of the wild-type (WT) and MUT TP53INP1 vectors used in the luciferase assay. C, The repression of luciferase activity by the TP53INP1 3′-UTR was dependent on miR-155. The MUT TP53INP1 3′-UTR abrogated the miR-155-mediated repression of luciferase activity. D, Expression of miR-155 after transfection of cardiac fibroblasts with pre-miR-155, anti-miR-155, or negative control (NC), as assessed by quantitative polymerase chain reaction (qPCR). E, Expression of TP53INP1 messenger RNA (mRNA) after transfection of cardiac fibroblasts with pre-miR-155, anti-miR-155, or NC, as assessed by qPCR. F, Western blot demonstrating TP53INP1 protein expression in cardiac fibroblasts treated with pre-miR-155, anti-miR-155, or NC. β-Actin was used as the loading control. The data are presented as the mean ± standard deviation (SD). *P < .05, **P < .01, and ***P < .001 versus control.
Moreover, ectopic expression of miR-155 was achieved by transfecting cardiac fibroblasts with pre–miR-155 or anti-miR-155 (Figure 5D). Quantitative real-time PCR assays showed that TP53INP1 mRNA expression levels of the pre–miR-155 group were lower than those of the NC group; in contrast, anti–miR-155 transfection upregulated TP53INP1 mRNA expression (Figure 5E). Furthermore, TP53INP1 protein expression levels were markedly downregulated in the pre–miR-155-transfected group and upregulated in the anti–miR-155-transfected group compared to the NC group (Figure 5F). These results indicate that miR-155 regulates TP53INP1 expression by directly targeting its 3′-UTR.
Downregulation of TP53INP1 Attenuates miR-155 KO-Mediated Inhibition of Cardiac Fibroblast Proliferation
To further elucidate the potential relationship between miR-155 and TP53INP1, we knocked down TP53INP1 using siRNA and then evaluated cell proliferation and related gene alterations in cardiac fibroblasts treated with TGF-β. We found that TP53INP1 expression was effectively decreased by siRNA in cardiac fibroblasts derived from WT or miR-155 KO mice (Figure 6A and B). Additionally, compared to the NC group, mRNA and protein expression levels of collagen I/III were dramatically upregulated in the si-TP53INP1 group, whereas the mRNA levels of caspase-3 and protein levels of cleaved caspase-3 were significantly downregulated (Figure 6C-F). Using an MTT assay, we found that TP53INP1 knockdown significantly promoted cell proliferation in the WT and miR-155 KO cardiac fibroblasts compared to the NC fibroblasts (Figure 6G), indicating that TP53INP1 knockdown attenuated the miR-155 KO-mediated inhibitory effects. Furthermore, TP53INP1 knockdown significantly increased the expression of α-SMA in the WT and miR-155 KO cardiac fibroblasts compared to the NC fibroblasts (Figure 6H). These results demonstrate that miR-155 regulates cardiac fibroblast proliferation and differentiation by targeting TP53INP1.

Knockdown of tumor protein p53-inducible nuclear protein 1 (TP53INP1) attenuates the effects of miR-155 knockout (KO) on transforming growth factor β (TGF-β)-induced cell proliferation and expression of collagen and cleaved caspase-3. The expression of TP53INP1 messenger RNA (mRNA; A) and protein (B) in cardiac fibroblasts derived from wild-type (WT) or miR-155−/− mice after 72 hours of TGF-β and TP53INP1 small interfering RNA (siRNA) treatment. The mRNA expression of collagen I (C), collagen III (D), and caspase-3 (E) in cardiac fibroblasts derived from WT or miR-155−/− mice after 72 hours of TGF-β and TP53INP1 siRNA treatment. F, Quantification of the protein expression of collagen I, collagen III, and cleaved caspase-3 in cardiac fibroblasts derived from WT or miR-155−/− mice after 72 hours of TGF-β and TP53INP1 siRNA treatment. G, Methylthiazol tetrazolium (MTT) assay-based measurements of cell proliferation in cardiac fibroblasts derived from WT or miR-155−/− mice after 72 hours of TGF-β and TP53INP1 siRNA treatment. H, Immunofluorescence staining for α-smooth muscle actin (α-SMA; red) with 4′,6-diamidino-2-phenylindole (DAPI) counterstaining (blue) in cardiac fibroblasts derived from WT or miR-155−/− mice after treatment with TGF-β and TP53INP1 siRNA (magnification, ×200). The data are presented as the mean ± standard deviation (SD). *P < .05, **P < .01, and ***P < .001 versus control. NC indicates negative control; si, siRNA; si-TP, TP53INP1 siRNA.
Discussion
In this study, we demonstrated that miR-155 expressed by cardiac fibroblasts is a potent contributor to AMI-mediated cardiac remodeling. Using gain-of-function and loss-of-function experiments in vivo, we demonstrated that miR-155 KO improved LV function, reduced infarct size, and attenuated collagen deposition, whereas the overexpression of miR-155 produced the opposite effects. Abnormal levels of miR-155 are related to inflammation, such as ischemia-induced inflammation. In one hand, microRNA-155 can mediate inflammatory response in ischemic tissues. 21 Recent studies showed that miR-155 KO in macrophages inhibited cardiac inflammation, hypertrophy, and failure in response to pressure overload. 22 Seok et al demonstrated that cardiac hypertrophy and cardiac remodeling could be suppressed in miR-155 null mouse hearts via targeting of jumonji, or adenine-thymine–rich interactive domain 2, in cardiomyocytes; this suppression prevented the progression of heart failure in response to transverse aortic restriction and calcineurin activation. 23 Interestingly, we found that miR-155 expression was significantly higher in cardiac fibroblasts, but not cardiac myocytes, after LAD ligation in WT mice. On other hand, ischemia induced by LAD ligation can release amount of inflammatory factors, such as tumor necrosis factor α and interleukin 6, which can activate miR-155 expression. 24 For example, lipopolysaccharides can induce miR-155 expression in macrophages, leading to the decrease in SMA. 25 It was reported that the serum levels of miR-155 were approximately 4-fold higher in patients who experienced cardiac death within 1 year after discharge. 26 Thus, there may be a positive feedback loop between miR-155 and inflammation. In this study, we found that AMI slightly upregulated miR-155 expression in myocytes but dramatically increased its expression in cardiac fibroblasts in WT mice, which may be activated by inflammatory factors. In addition, we found that AMI downregulated TP53INP1 expression in cardiac fibroblasts, but not in myocytes, in WT mice, indicating there is a negative relationship between miR-155 and TP53INP1. Thus, miR-155 KO in cardiac fibroblasts may be able to potently improve cardiac remodeling.
In addition, Kishore et al reported that intramyocardial delivery of bone marrow-derived progenitor cells to infarcted diabetic db/db mice inhibited miR-155 expression, leading to the repression of profibrotic signaling in cardiac fibroblasts; this effect was protective for cardiac fibrosis and function. 27 In the other types of fibrotic diseases, such as lung fibrosis, primary myelofibrosis, and lumbar spinal stenosis, miR-155 expression levels are positively correlated with the degree of fibrosis. 28 –30 A recent study illustrated that local downregulation of miR-155 expression at skin wound edges clearly disrupted immune cell requirements, reduced proinflammatory factors and the expression of α-SMA and collagen I/III at both the mRNA and protein levels, and importantly did not significantly alter the rate of healing. 31 In this study, we stimulated cardiac fibroblasts isolated from the hearts of WT, miR-155+/+, and miR-155−/− mice using TGF-β, a pleiotropic cytokine that promotes fibrosis. 32 Our results revealed that TGF-β treatment induced the expression of miR-155, triggered the upregulation of collagen I/III, and reduced the expression of cleaved caspase-3 in a time-dependent manner in cardiac fibroblasts. These effects were enhanced by miR-155 overexpression and diminished by miR-155 KO. In addition, TGF-β–induced cardiac fibroblast proliferation was blocked by miR-155 KO. Moreover, miR-155 KO also dramatically decreased the expression of α-SMA, indicating a blockade of cardiac fibroblast differentiation into myofibroblasts. 33,34 Activated myofibroblasts secrete fibrogenic growth factors, such as TGF-β, which lead to collagen synthesis and fibrosis and, finally, progressive adverse myocardial remodeling. 35,36 Taken together, our results provide evidence that KO of miR-155 plays an important role in the improved LV function and cardiac remodeling that is associated with a reduction in fibrosis. 22
Our study suggests that the protective effects of miR-155 KO on cardiac remodeling are mediated at least in part by TP53INP1. We found that downregulation of TP53INP1 rescued blunted fibroblast proliferation and differentiation into myofibroblasts. Tumor protein p53-inducible nuclear protein 1 is a proapoptotic, stress-induced p53 target gene that has the ability to interact with p53 and modulates its transcriptional activity. In turn, p53 is also capable of activating TP53INP1 transcription. 37 Overexpression of TP53INP1 induces cell cycle arrest and apoptosis in several cell lines, including fibroblasts. 38 Tumor protein p53-inducible nuclear protein 1 has been identified as a target of miR-155 in several tumors, including pancreatic tumors 39 as well as in breast cancer 40 and liver cancer. 41 Using a luciferase report assay, we determined that miR-155 directly targets the 3′-UTR of TP53INP1 in cardiac fibroblasts and regulates its expression at both the mRNA and protein levels. Furthermore, the loss of TP53INP1 expression reversed the inhibitory effects of miR-155 KO on the effects of TGF-β treatment on collagen I/III expression and fibroblast proliferation and differentiation. A previous report found that TGF-β could indirectly regulate TP53INP1 expression via miR-155 in liver cancer cells. 42 Altogether, these data suggest that miR-155 KO induces the upregulation of TP53INP1, leading to inhibition of cardiac fibroblast proliferation and a reduction in myofibroblasts, which improves cardiac remodeling.
In conclusion, using loss-of-function and gain-of-function experiments in vivo and in vitro, we illustrated that the inhibition of miR-155 in cardiac fibroblasts prevents cardiac remodeling by targeting TP53INP1. Despite the complex process that underlies cardiac remodeling, our data suggest that miR-155 may be a therapeutic target for cardiac remodeling induced by AMI.
Footnotes
Author Contributions
W. He and H. Huang contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted the manuscript, critically revised the manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy. Q. Xie contributed to analysis, critically revised the manuscript, and agrees to be accountable for all aspects of work ensuring integrity and accuracy. Z. Wang contributed to acquisition and interpretation. Y. Fan contributed to analysis. B. Kong contributed to acquisition. D. Huang contributed to acquisition and analysis. Y. Xiao contributed to acquisition and interpretation and critically revised the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Key Project of Hubei Science and Technology Support Program (No. 2013BCB013), the Key Project of Hubei Natural Science Foundation (No. 2013CFA059), and the National Natural Science Foundation of China (No. 81270249).