
Abstract
Dyslipidemia is a major risk factor for the development of coronary artery disease, a leading cause of morbidity and mortality worldwide. Lowering low-density lipoprotein (LDL) has significantly reduced the risk of death and other major cardiovascular events, and statins remain the therapy of choice. However, as some patients are limited by the side effects of statins, cannot achieve their target LDL on statin therapy, or have other abnormalities in their lipid profile, alternative agents are being developed. In this review, we highlight the major classes of novel nonstatin LDL-lowering agents that are currently in various stages of development. Although many hold great promise, the results of large Phase III trials will be needed to definitely establish the efficacy, safety, and clinical utility of these agents in the general population.
Keywords
Introduction
Lowering low-density lipoprotein (LDL) cholesterol (LDL-C) continues to remain one of the most important treatment goals in the prevention of atherosclerosis and its complications, including acute coronary syndromes (ACS) in patients with hypercholesterolemia. 1,2 Many recent studies and meta-analyses have continued to highlight the relationship that exists between low achieved LDL concentrations and decrease in adverse clinical outcomes, 3 in both primary 4 and secondary prevention cohorts. 5 Benefit is positively correlated with the degree of LDL lowering, although there is an ongoing debate whether decreasing LDL from an already low achieved value further decreases clinical events. It appears that very low concentrations of LDL (<40 mg/dL) are relatively safe, without an increase in the risk of adverse effects related to the medications needed to achieve these levels 6 ; however, additional outcomes data will be needed to definitively determine what the lower limit of LDL is at which there is no incremental lowering of risk.
Statins, the first-line agents to treat hyperlipidemia, can effectively lower LDL significantly and are well tolerated. Despite the widespread use of statins and other lipid-lowering therapy, however, there remain a substantial number of patients at risk who are still unable to achieve their lipid targets despite taking maximally tolerated statin therapy 7,8 and many remain undertreated with statins following an ACS, 9 possibly due to the side effects of these medications. One estimate reports that 71 million people in the United States with hyperlipidemia and 12 million people in Europe have lipids that are uncontrolled, and 1.4 million of these patients are “intolerant” or refractory to statins 10 ; in Europe, as many as one-half of the patients are untreated. 11,12 For this reason, alternative highly potent LDL-C lowering medications are desirable as monotherapy as well as adjunctive therapies for patients with hypercholesterolemia who are in need of LDL lowering.
Improvements in molecular biology techniques, genetic sequencing, and drug discovery have led to the investigation of a number of novel targets for lowering LDL and the development of a variety of new compounds designed to interrupt different steps within the pathway of cholesterol metabolism. In this review, we highlight the major classes of novel compounds that are currently under development to lower LDL and improve lipid profiles (Table 1), focusing on ongoing clinical investigational data, efficacy, and adverse effects.
Major Novel LDL-C Lowering Agents.

Abbreviations: Apo-AI, apolipoprotein AI; Apo-B, apolipoprotein B; ALT, alanine transaminase; AT, aminotransferase; BP, blood pressure; CK, creatine kinase; CRP, C-reactive protein; HDL-C, high-density lipoprotein cholesterol; HR, heart rate; LDL-C, low-density lipoprotein; LDL-R, low-density lipoprotein receptor; LFT, liver function test; Lp(a), lipoprotein (a); PCSK9, proprotein convertase subtilisin/kexin type 9; PO, orally; qd, once daily; SAE, serious adverse events; SC, subcutaneously; TGs, triglycerides; ULN, upper limits of normal; >3×, more than 3 times; >2×, more than 2 times; TBL, total billirubin.
Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitors
First discovered as a regulator of cholesterol homeostasis, proprotein convertase subtilisin/kexin type 9 (PCSK9) was initially identified as the gene product of the third genetic locus associated with autosomal dominant hypercholesterolemia. In the HC92 family pedigree, family members with gain of function mutations in this gene had total cholesterol in the 90th percentile, tendon xanthomas, coronary disease, and premature myocardial infarction (MI) and stroke. 34 These discoveries led to better characterization of the structure and mechanism of action of PCSK9 (Figure 1). Synthesized as a proprotein in hepatocytes, PCSK9 undergoes self-cleavage and activation and is secreted into the serum. It then binds to the epidermal growth factor domain of the LDL receptor (LDL-R) and is endocytosed along with the LDL-R and bound LDL particle. Within the endosome, PCSK9 targets the LDL-R protein for catalytic degradation, preventing its recycling to the cell surface which hinders its ability to scavenge freer LDL that is circulating in the serum. 36 –38 If PCSK9 is inhibited, however, the LDL-R is able to return to the cell surface and remove more circulating LDL-C from the blood, effectively lowering the concentration of circulating LDL-C.

PCSK9 exerts its effects by binding to LDL-R and targeting it for endosomal degradation, preventing it from being recycled to the hepatocyte surface to scavenge more circulating LDL-C. By inhibiting PCSK9, either via monoclonal antibody or by inhibiting protein synthesis using a siRNA, more LDL-R can be recycled, allowing for more uptakes of LDL and a decrease in serum concentration. Adapted with permission from Garber, 35 © 2012 Nature Publishing Group, a division of Macmillan Publishers Limited. LDL-R indicates low-density lipoprotein receptor; PCSK9, proprotein convertase subtilisin/kexin type 9; LDL-C, low-density lipoprotein cholesterol; siRNA, small interfering RNA.
Following its initial discovery, transgenic animal models were generated in which adenoviral-mediated overexpression 39 or knockout mutations in PCSK9 36 significantly decreased or increased LDL-R expression, respectively, and increased or decreased LDL-C, respectively. Sequence variations identified in black individuals with nonsense mutations in PCSK9 were associated with reductions in LDL-C by 28% to 40% and lower incidence of coronary heart disease by 88% (P = .008). 40,41 This generated great interest in inhibition of this protein as a target to improve outcomes. Furthermore, PCSK9−/−mice had a more robust response to statin therapy (suggesting that PCSK9 inhibition and statin therapy may be synergistic 39 ), raising expectations even more for this class of drugs.
Several physiological changes occur in the setting of statin therapy, including an upregulation of PCSK9 as well as LDL-R expression by the hepatocyte. 42 –44 This has been proposed as a plausible mechanism limiting the effectiveness of statins, especially at higher doses. However, upregulation of PCSK9 also appears to explain heightened responsiveness to PCSK9 inhibition in the setting of statin therapy. Therefore, by inhibiting the physiological mechanism of statin resistance, PCSK9 inhibitors can presumably work synergistically with statins to more effectively lower LDL-C than statins alone.
PCSK9 can be inhibited by a variety of mechanisms including, but not limited to, monoclonal antibodies and RNase H antisense inhibitors. RNase H inhibitors are still early in development, with data from animal studies showing a 92% decrease in PCSK9 messenger RNA (mRNA), a 2-fold increase in hepatic LDL-R, and a 38% decrease in LDL-C. 45 There is also ongoing research to develop shorter, more potent antisense oligonucleotides to inhibit target mRNAs. 46 In contrast, there are multiple monoclonal antibodies against PCSK9 in clinical trials and 2 forerunners that are currently being tested in large, outcomes-based Phase III trials.
Recombinant Monoclonal Antibodies
Fully human monoclonal antibodies have the greatest specificity for PCSK9 and target extracellular circulating PCSK9 most effectively. Because of their mechanism of action as antibodies and concerns regarding stability of the protein, this class of drugs cannot be formulated orally and must be solubilized and prepared for injection. Four fully human recombinant monoclonal antibodies (REGN727/SAR236553 [Regeneron Pharmaceuticals, Tarrytown, New York]; AMG145 [Amgen Pharmaceuticals, Thousand Oaks, California]; RN316 (PF-04950615) [Pfizer, New York, New York]; RG7652 [Roche, Indianapolis, Indiana]) showed promising results in early phase studies 47 –50 (see below), and SAR 236553 and AMG145 have started large Phase III trials in 2012 and 2013. 21,22,25,51 The mode of administration (subcutaneous injection) of SAR 236553 and AMG145 has been well tolerated in Phase II trials, 21,22,25 and no treatment-related adverse events have been clearly established in the modest number (<3000) of patients treated for 12 weeks to date. The efficacy of these antibodies has been consistent across diverse patient populations and appears additive, rather than synergistic, to statins.
SAR236553/REGN 727
In healthy volunteers, escalating doses of SAR236553 reduced LDL by up to >60% and lasted for at least 30 days in patients administered with higher doses. 23 A comprehensive Phase I program was conducted and consisted of multiple patient populations: healthy patients (n = 72), heterozygous familial hypercholesterolemia (FH; n = 21), non-FH (n = 30), with/without statins, and multiple doses and routes of administration (intravenous and subcutaneous) and established dose-dependent decreases in LDL-C. 21 An LDL-C reduction of 61% occurred at the maximum dose of 150 mg SAR236553/REGN 727. 21 There were no major serious adverse effects and no patients discontinued drug due to adverse effects.
A Phase II trial randomized 183 patients with LDL-C of ≥100 mg/dL on a stable statin dose to either placebo or 1 of the 4 active dose regimens of SAR236553 (50 mg, 100 mg, or 150 mg every 2 weeks; or 200 mg or 300 mg every 4 weeks). 22 The regimen of 150 mg SAR236553 every 2 weeks was the most efficacious dose regimen studied decreasing LDL-C by 72% and achieving an LDL-C target of <70 mg/dL in 100% of the recipients. 22 Interestingly, in addition to LDL-C, there was a dose-dependent decrease in apolipoprotein-B (ApoB), triglycerides (TGs), and lipoprotein(a) (Lp(a)). There was a single case of leukocytoclastic vasculitis in a patient who received the maximal dose of the drug (300 mg SAR236553). The vasculitis was resolved with steroids, and although it was considered unrelated to therapy, it does raise the question of immunogenicity of these compounds. At the 20-week follow-up, minimally detectable antidrug antibodies were found. An open-label Phase II extension study is ongoing (NCT01576484). In another study of patients with hypercholesterolemia, the use of SAR236553 in patients administered with either 10 mg or 80 mg atorvastatin resulted in a significantly greater reduction in LDL-C than that attained with atorvastatin alone (73% vs 17%). 48 In November 2012, the start of the ODYSSEY outcomes trial (NCT01644474) was announced, in which approximately 18 000 patients with a recent ACS who are not at their LDL-C goal on maximal statins will be randomized to SAR236553, 75 mg once every 2 weeks (Figure 2).

Trial overview of the ongoing Phase III clinical trial ODYSSEY, which is randomizing patients after acute coronary syndrome to the PCSK-9 antibody SAR236553 versus placebo on a background of evidence-based medical therapy. The trial is targeted to enroll approximately 18 000 patients with an estimated follow-up of 6 years. ACS indicates acute coronary syndrome; SQ, subcutaneously; CHD, coronary heart disease; NFMI, non-fatal myocardial infarction; UA→hospitalization, unstable angina leading to hospitalization; NF, nonfatal; PCSK9, proprotein convertase subtilisin/kexin type 9.
This antibody has also demonstrated efficacy in other populations, such as patients with FH. In a Phase II study comprising 77 of such patients on a stable statin dose, 150 mg SAR236553 administered every 2 weeks lowered LDL-C by up to 90% with no increase in hepatic transaminases or creatine kinase, 21 providing a highly effective adjunctive treatment for this otherwise refractory population.
AMG145
Another fully human monoclonal antibody (AMG 145), with a mechanism of action that is similar to SAR236553, is currently under development. Phase I data in healthy volunteers (Phase Ia) as well as those on a background of statin therapy (Phase Ib) demonstrated a dose-dependent decrease in LDL-C and unbound PCSK9, with LDL-C reductions of up to 81% at maximal doses over and above background statin therapy 25 without any serious apparent adverse events or discontinuations related to adverse events. A comprehensive Phase II program for AMG145 was undertaken (1) on a background of lipid-lowering therapy in patients with hypercholesterolemia (DESCARTES [full trial names provided at end of article]; NCT01516879; n = 905); (2) in combination with statins in patients with hypercholesterolemia: (LAPLACE TIMI 57; n = 631) 47 ; (3) as primary prevention/monotherapy in low-risk populations (MENDEL; n = 406) 50 ; (4) in heterozygous FH (RUTHERFORD; n = 168) 52 ; (5) as monotherapy in patients with statin intolerance (GAUSS; n = 160) 53 ; (6) in homozygous FH (NCT01588496; n = 67); (7) as a long-term open-label extension safety and tolerability study of AMG145 (OSLER; NCT01439880; n ∼ 1400).
In the largest Phase II study of this compound to date, LAPLACE-TIMI 57, ascending doses and 2 dosing frequencies of AMG 145 in 631 patients with hypercholesterolemia on statin therapy were tested in a randomized fashion. 54 AMG145 resulted in the reduction of LDL-C to 42% to 66% at the end of the dosing interval without any apparent treatment-related adverse events or drug antibodies. 47 The LDL-C was reduced by up to 85% 1 week after dosing. Among the 284 high-risk patients in this study, 90% were able to achieve LDL-C <70 mg/dL with higher doses of AMG145. In addition, there was a 23% to 30% decrease in Lp(a) observed with higher doses of AMG145 47 in all patients, suggesting that PCSK9 inhibition may have multiple mechanisms for improving cardiovascular outcomes.
The PCSK9 inhibition has demonstrated consistent results in all populations: as monotherapy in MENDEL 50 (LDL-C lowered by 48% to 51% reduction and Lp(a) lowered by 30%); in patient with statin intolerance in GAUSS 53 (LDL-C lowered by 41% to 51% and up to 63% with eztemibe); and in patients with heterozygous FH in RUTHERFORD 52 (LDL-C lowered by 43% to 55%).
RN316 (PF-04950615)
A third PCSK9 immunoglobulin G antibody (RN316 or PF-04950615) being developed by Pfizer has completed Phase I (NCT00991159; NCT01163851) and one Phase II trial (NCT0135014). In the first half of the Phase I program, RN316 was administered intravenously to 48 healthy participants aged 18 to 70 years, in escalating doses (0.3 mg/kg, 1.0 mg/kg, 3.0 mg/kg, 6.0 mg/kg, 12.0 mg/kg, and 18.0 mg/kg) to assess safety and tolerability. In the second half of the Phase I program, a single dose of RN316 was administered to the patients with hypercholesterolemia on a stable dose of atorvastatin (40 mg) to assess pharmacokinetics and pharmacodynamics of the drug. 51 The results of the Phase II program (n = 135) demonstrated an LDL-C reduction of 46% and 56% at the highest doses of 3 mg/kg and 6 mg/kg, respectively. 51 RN 316 had to be interrupted due to LDL-C <25 mg/dL, as specified by the protocol. Those patients who received all 3 doses of RN316 at high doses had reductions in LDL of approximately 75%. There were 5 reports of myalgias, and 5 patients had an increase in creatine kinase. There were, however, drug antibodies detected in 5 of the 118 patients, although these antibodies did not affect the efficacy of the drug or have other clinical effects.
RNase H Antisense Inhibitors
These single-stranded (DNA or RNA) nucleotides are complementary to a mRNA sequence and prevent protein synthesis by binding and preventing translation of the mRNA. 46 Two sponsors, Isis/Bristol-Myers-Squibb (BMS-844421; NCT01082562) and Santaris Pharmaceuticals (SPC5001; NCT01350960), halted Phase I development of their agents, prematurely terminating trials, presumably due to safety issues. Although detailed results of the study have not yet been published, Alnylam Pharmaceuticals, another sponsor developing a drug in this class, announced in early January 2012 that ALN-PCS was able to reduce the production of the PCSK9 gene product by 66% and was found to be safe and well tolerated in Phase I (NCT01437059). A dose-ranging Phase II study is planned.
Cholesteryl Ester Transfer Protein Inhibitors
Although initially designed with the primary intent of raising high-density lipoprotein (HDL) cholesterol (HDL-C), some inhibitors of the cholesteryl-ester transfer protein (CETP) also lowered LDL-C. Thus, we will discuss in the following section CETP inhibitors that are still in development and have a robust LDL-C lowering effect.
CETP is intimately involved in the process of reverse cholesterol transport (transferring cholesterol from the tissues to the liver) by mediating transfer between the lipoproteins. When extrahepatic cells remove excess cholesterol and transfer it as free cholesterol to HDL, it is subsequently delivered to the liver or converted into cholesteryl esters (CEs). These CEs are then transported to the liver by a direct pathway or indirectly via LDL or very-LDL (VLDL). The transport of CEs from HDL to LDL/VLDL occurs via CETP (Figure 3). Inhibition of CETP allows the CEs to remain within the HDL particle or get transported directly to the liver, without being first transferred to LDL/VLDL.

Reverse cholesterol transport is a multistep process in which cholesterol from peripheral tissues and vessel wall returns to the liver. Shown here is a simplified schematic of that process. Free cholesterol is transferred to HDL via the ABC-G1 and ABC-A1 transporters. After conversion to CEs by enzymatic conversion, the CEs can be transferred to other lipoproteins (including LDL) in exchange for triglycerides by CETP. Inhibition of CETP can inhibit this exchange, resulting in lower circulating CE-rich LDL particles. LDL indicates low-density lipoprotein; VLDL, very-low-density lipoprotein; HDL, high-density lipoprotein; ApoB, apolipoprotein B; CE, cholesteryl ester; Free Chol, free cholesterol; CETP, cholesteryl ester transfer protein; ABC-G1/-A1, ATP binding cassette transporter-G1/-A1. Courtesy: Christopher P. Cannon, MD.
The first 2 CETP inhibitors studied in large phase III outcomes trials (torcetrapib 55 and dalcetrapib 56 ) did not reduce cardiovascular events, and in fact torcetrapib was associated with an increase in mortality. 55 An increase in adverse effects was thought to be mediated by off-target actions of torcetrapib, including increases in aldosterone, serum sodium, and systolic blood pressure, likely mediated via unintentional effects on adrenal cells. 57 The effects of torcetrapib on adrenal cells do not appear to be a class effect, and several newer agents that were developed later (anacetrapib, evacetrapib) do not demonstrate such off-target effects 57 yet do robustly lower LDL-C.
Anacetrapib
Anacetrapib, a very potent and reversibly binding CETP inhibitor, is now the forerunner in this class. In the phase II study (DEFINE), 1623 patients were randomized to receive 100mg of anacetrapib versus placebo for 18 months. The primary efficacy outcome was percentage change from baseline in LDL-C at 24 weeks (HDL-C was a secondary outcome) and safety outcomes were assessed through 76 weeks. There was a robust LDL lowering (40% based on calculated LDL-C; 25% to 35% based on measured LDL-C) and HDL raising (138%) over placebo without any of the adverse effects on blood pressure, electrolytes, and aldosterone seen with torcetrapib. 13 Based on a prespecified Bayesian analysis in this study, these results provided a 94% confidence that anacetrapib would not be associated with a 25% (or greater) increase in cardiovascular events, as was seen with torcetrapib. In the ongoing Phase III trial of anacetrapib, REVEAL-HPS3 TIMI 55, approximately 30 000 patients aged 50 years or older with preexisting occlusive vascular disease and LDL-C of <100 mg/dL are being randomized to atorvastatin plus 100mg anacetrapib daily or atorvastatin plus placebo with a primary end point of any major coronary event (coronary death, nonfatal MI, or coronary revascularization).
Evacetrapib
Evacetrapib is a fourth CETP inhibitor that has been developed and recently completed a Phase II trial. 58 Multiple doses of evacetrapib (30mg, 100mg, and 500mg per day) demonstrated that monotherapy with this CETP inhibitor lowers LDL by 20% to 51% in a dose-dependent fashion compared to placebo and is additive to background statin for the lowering of LDL by 11% to 14% (P <.001). 19 Similarly, HDL-C was increased with evacetrapib in a dose-dependent fashion by 55% to 129%, but there was no synergy seen in the presence of statin (P = .39). A large Phase III clinical outcomes trial with evacetrapib (ACCELERATE; NCT 01687998) is ongoing and will randomize approximately 11 000 high-risk patients with vascular disease to evacetrapib 130 mg once daily versus placebo and follow patients for about 4 years. The primary outcome measure will be time to first occurrence of the composite end point of cardiovascular death, MI, stroke, coronary revascularization, or hospitalization for unstable angina.
Other CETP inhibitors
There is currently another CETP inhibitor, BAY 60-5521, which was clinically safe and well tolerated without effects on heart rate, blood pressure, and electrocardiogram recordings that were observed during the first-in-man study. Further development is being pursued. 20
ETC-1002: Decreasing LDL-C by Decreasing Its Synthesis
Fatty acid and cholesterol synthesis occurs in the liver via multistep enzymatic pathways. ETC-1002 is a once daily, small molecule oral inhibitor of both cholesterol and fatty acid synthesis. 59 By inhibiting adenosine monophosphate (AMP)-activated protein kinase, it decreases the cyclic AMP-mediated downstream pathways, including cholesterol synthesis and oxidation of fatty acids, resulting in lower synthesis of LDL-C. 60 In addition to decreasing LDL-C, this agent also regulates other metabolic imbalances, such as aberrations in glucose homeostasis, making it an ideal candidate for those patients who have or are at risk of developing metabolic syndrome. In animal models, ETC-1002 decreased cholesterol and TG concentrations and hepatic glucose output, thereby reducing body weight, decreasing inflammation, and reducing hepatic steatosis. 61
In a Phase I study of this compound (NCT01485146), 71 healthy patients were administered with ETC-1002 daily. 26 The LDL-C was reduced by day 5, and the nadir was reached by day 15 with LDL-C reduction of up to 27%. Results were similar in patients with mild dyslipidemia. There were no dose-limiting adverse effects, no concerning laboratory abnormalities, and only one patient withdrew from the study for personal reasons. A Phase II (NCT01262638) randomized dose-finding placebo-controlled study was carried out using 3 doses (40mg, 80mg, and 120mg) in patients with TGs of <150 mg/dL and patients with TGs of ≥150 mg/dL once daily for 12 weeks. 27 In all patients, regardless of baseline TG level, there was a dose-dependent reduction in LDL-C at 12 weeks, ranging from 18% at the lowest dose (40mg) to 24% to 29% at the highest dose (120mg). There was also a qualitatively similar dose-dependent decrease in ApoB and LDL particle number. 27 Although there was no significant change in HDL-C, there was a U-shaped dose–response with respect to HDL particle number by nuclear magnetic resonance spectroscopy. ETC-1002 provides an appealing agent that not only reduces LDL-C but also improves the metabolic profile of patients at risk of metabolic syndrome. However, additional safety and outcomes data are needed prior to its widespread clinical use.
The ApoB-100 Inhibitors
The ApoB is the primary structural and transport protein of LDL-C. Because of its integral role in LDL function and structure, ApoB provides an appealing target for targeting LDL-C and modulating cholesterol delivery to cells. With evolving molecular techniques, single-stranded antisense oligonucleotides, which bind to a very specific mRNA sequence and target this mRNA for degradation, have been developed corresponding to ApoB-100 62 (Figure 4).

Mechanism of action of mipomersen. Mipomersen is a novel antisense oligonucleotide which inhibits the transcription and synthesis of apolipoprotein-B100, thereby leading to less functional LDL. Adapted with permission from Kohli and Cannon, 63 © European Society of Cardiology, Oxford University Press. LDL indicates low-density lipoprotein.
Mipomersen (Genzyme, Cambridge, Massachuttes) is a second-generation small molecule oligonucleotide that is administered as a subcutaneous injection. 46 It has a long half-life, and has dual metabolism, in both the liver and the kidneys. In Phase I (NCT01061814) 64 and Phase II studies (NCT00707746), 65 ApoB was decreased with mipomersen, and there was a significant (∼30%-50%) reduction in LDL-C and Lp(a). However, these effects were accompanied by elevations in transaminases and injection site reactions. 64,66 In a small study of high-risk patients on a stable dose of statin, 74 patients were randomized in a 4:1 fashion to receive either once weekly mipomersen (200mg) subcutaneously or placebo. Most lipid parameters were favorably affected, including reductions in LDL-C (−21% to −52%) and ApoB (−19% to −54%). 65 Similar results were seen in a statin-intolerant cohort (n = 34) 66 and patients with homozygous FH already on potent statins. 26 The drug has thus far been tested in 4 Phase III trials, 3 of which included patients with severe FH or hypercholesterolemia and 1 which targeted those with risk factors for coronary disease. 46 The pooled data, consisting of 261 individuals with FH, reported a reduction in LDL-C from baseline to week 26 ranging from 25% to 37%. In the study with 34 patients with homozygous FH, 25% reduction in LDL-C was seen with an overall treatment effect of 22% compared with placebo. However, interindividual responsiveness was highly variable with a range of +2% to −82% in the LDL-C observed.
Despite these favorable results, adverse effects were common and clinically troublesome and therefore may limit the clinical use of mipomersen. Adverse effects included transaminitis, hepatic steatosis, flu-like symptoms, injection site reactions, proteinuria, and inflammatory reactions in the liver. In the pooled Phase III studies, 8% of mipomersen-treated patients had liver function tests more than 3 times the upper limits of normal and dosing with mipomersen was stopped in approximately 5% of the patients due to transaminitis. There was also a significant proportion of the mipomersen group (62%) that had an increase in hepatic fat content of ≥5%, compared with 8% in the placebo group. However, those that had liver biopsies had minimal or no inflammation and fibrosis present despite the steatosis, and very few (0.3%) patients had persistence of steatosis 24 weeks after the last dose of mipomersen, 61 suggesting that the fat accumulation is reversible. The number of injection site reactions was also imbalanced between mipomersen and placebo arms (84% vs 33%), leading to drug discontinuation in 5% of mipomersen-treated patients. There were also concerns that emerged about flu-like reactions, increase in inflammation (as measured by transient increases in C-reactive protein), anti-mipomersen antibodies, and proteinuria associated with mipomersen. 29
Despite these adverse effects, in October 2012, the Food and Drug Administration (FDA) approved this agent for patients with homozygous FH (who are notoriously difficult to treat) already on a statin. Given the safety profile of these agents, however, antisense ApoB-100 inhibitors are not likely to be frontline adjunctive agent in patients with hypercholesterolemia unless a better safety profile can be demonstrated. 63
Microsomal TG Transport Protein Inhibitors
Microsomal TG transport protein (MTP) is located both on hepatocytes and on enterocytes and participates in generation of VLDLs and chylomicrons from cholesterol, TGs, and ApoB. Inhibitors of this intracellular protein exert a dual mechanism of action by blocking cholesterol absorption in the intestines as well as hepatic secretion of VLDL (Figure 5). In early studies, MTP inhibitors (CP-346086, AEGR-733, and BMS-201038) were shown to lower LDL-C by 51% and ApoB by 56% but resulted in a large number of patients with elevation in liver function tests and hepatic steatosis due to excess accumulation of TGs within the liver. 32 Furthermore, there was an increased incidence of gastrointestinal side effects observed, especially a transient mild to moderate increase in stool frequency.
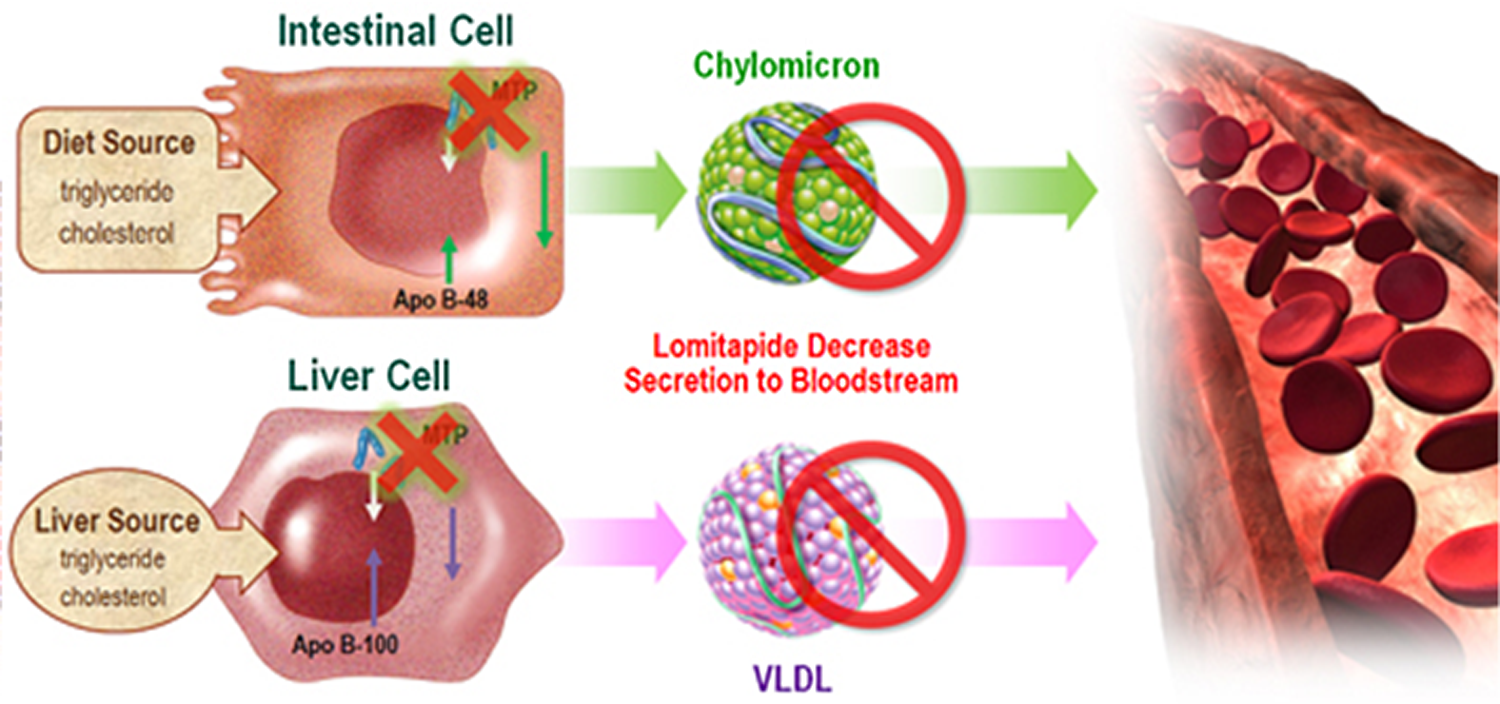
Mechanism of action of microsomal triglyceride transport protein inhibitor. There are dual sites of action, (1) enterocyte and (2) hepatocyte. Adapted with permission from Aegerion, 67 © Aegerion Pharmaceuticals, Inc. MTP indicates microsomal triglyceride transport protein; ApoB, apolipoprotein B; Free Chol, free cholesterol; LDL, low-density lipoprotein; VLDL, very low-density lipoprotein.
Based on its mechanism of action of inhibiting efflux of TGs from the liver, the adverse effects associated with these drugs were presumed to be dose dependent and a Phase II study with significantly lower doses were undertaken. After 12 weeks of AEGR-733 monotherapy (n = 84) in a Phase II study, there was a dose-dependent reduction in LDL-C ranging from 19% at the lowest dose to 30% at the highest. 30 AEGR-733 was studied as monotherapy as well as an adjunct to ezetemibe; since it exerts its mechanism of action in the intestines, it is similar to ezetemibe and may be additive or synergistic. Indeed, when combined with ezetemibe, there was an additive effect, with a decrease of 46% in LDL-C, when compared to only 20% with ezetemibe alone.
Although the LDL-C reduction was in a range that would translate into clinical benefit, there was a small decrease in HDL-C (7%), modest weight loss (possibly due to fat malabsorption), and high rate of discontinuation (32% [9 of 28] of the patients who were on monotherapy and 14% [4 of 28] of those on AEGR-733 and ezetemibe combination therapy). The most common reason for discontinuation was mild transaminase elevations that occurred in 18% (9 of 56) on AEGR-733, either alone or in combination with ezetemibe. Given their tolerability and safety profile, MTP inhibitors would appear to have a limited role, being used mostly in high-risk patients who are statin-intolerant or have homozygous FH. Notably, this agent was also approved by the FDA in October 2012 as an adjunct to a low-fat diet, other drugs, with or without apheresis in patients with homozygous FH.
Summary
There are a number of novel nonstatin drugs in various phases of development that lower LDL-C. Although statins will continue to remain first-line therapy, these novel therapeutic classes provide adjunctive treatments for patients with statin intolerance and/or those who are not achieving their lipid targets. Of these agents, PCSK9 inhibitors and the second-generation CETP inhibitors hold the most promise due to their potent effects on lipoproteins and excellent tolerability thus far. Phase III trials with PCSK9 monoclonal antibodies, which decrease LDL up to 72% and have shown efficacy in multiple patient populations, are ongoing. Although these monoclonal antibodies offer potent new therapies for lowering LDL, there is a theoretical concern regarding their immunogenicity, including the development of neutralizing and nonneutralizing antibodies against these agents. Anacetrapib, a more potent CETP inhibitor that also lowers LDL-C by >30%, is being evaluated in a Phase III study comprising 30 000 patients, and results are expected in the next 4 to 5 years. Other drugs targeting diverse steps in cholesterol synthesis and structure are highly efficacious. The ApoB 100 inhibitors and MTP inhibitors appear to be limited in their clinical application due to adverse effects and toxicity, with most of these adverse effects related to liver function abnormalities. Furthermore, due to the nature of such targeted therapies, they have to be formulated as subcutaneous injections and may have limited oral bioavailability. This may limit the widespread adoption of such therapies. Nonetheless, these other drugs may offer physicians with potent therapies (after statins and ezetemibe/bile acid resins) for adjunctive treatment or as alternatives in patient with statin intolerance with high LDL-C. Importantly, the new molecular targets for lowering LDL-C have opened the door to the introduction of inhibitory antibodies, RNA, and small molecules into the world of cardiovascular therapeutics.
Footnotes
Abbreviations
Declaration of Conflicting Interests
The author(s) declared a potential conflict of interest as follows: Both authors participated in the LAPLACE-TIMI 57 trial sponsored by Amgen. Dr Kohli has served on the advisory board for Daiichi-Sankyo. Dr Giugliano has received honoraria for consulting from Amgen, Regeneron, and Sanofi.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.