
Abstract
We recently showed that Bendavia, a novel mitochondria-targeting peptide, reduced infarction and no-reflow across several experimental models. The purpose of this study was to determine the therapeutic timing and mechanism of action that underlie Bendavia’s cytoprotective property. In rabbits exposed to in vivo ischemia/reperfusion (30/180 min), Bendavia administered 20 minutes prior to reperfusion (0.05 mg/kg/h, intravenously) reduced myocardial infarct size by ∼50% when administered for either 1 or 3 hours of reperfusion. However, when Bendavia perfusion began just 10 minutes after the onset of reperfusion, the protection against infarction and no-reflow was completely lost, indicating that the mechanism of protection is occurring early in reperfusion. Experiments in isolated mouse liver mitochondria found no discernible effect of Bendavia on blocking the permeability transition pore, and studies in isolated heart mitochondria showed no effect of Bendavia on respiratory rates. As Bendavia significantly lowered reactive oxygen species (ROS) levels in isolated heart mitochondria, the ROS-scavenging capacity of Bendavia was compared to well-known ROS scavengers using in vitro (cell-free) systems that enzymatically generate ROS. Across doses ranging from 1 nmol/L to 1 mmol/L, Bendavia showed no discernible ROS-scavenging properties, clearly differentiating itself from prototypical scavengers. In conclusion, Bendavia is a promising candidate to reduce cardiac injury when present at the onset of reperfusion but not after reperfusion has already commenced. Given that both infarction and no-reflow are related to increased cellular ROS, Bendavia’s protective mechanism of action likely involves reduced ROS generation (as opposed to augmented scavenging) by endothelial and myocyte mitochondria.
Introduction
Early and successful reperfusion of the acutely ischemic myocardium is the most effective salvage against irreversible ischemic injury (infarction) but can also cause reperfusion injury. Among the cellular culprits that contribute to reperfusion injury, bursts of reactive oxygen species (ROS) occurring during early reperfusion have been known to occur since the late 1980s. 1,2 Transient production of ROS contributes to electromechanical dysfunction, infarction, and the coronary no-reflow phenomenon (reviewed in 3,4 ). Despite a clear understanding of the role of ROS in reperfusion injury, there are no adjuvant treatments currently administered in routine clinical practice to protect the heart against reperfusion injury.
The myocardial milieu is a hostile environment in early reperfusion. Increased ROS levels, aberrant cellular calcium handling, and restoration of cytosolic pH activate/open mitochondrial pores and decrease bioenergetic capacity, 5 –9 often inducing necrotic and apoptotic cell death. 10 Scores of strategies have attempted to mitigate injury by targeting ROS and/or by blocking mitochondrial pores, but few have translated into clinical practice. 11 –13 The National Institutes of Health has recently convened workshops seeking suggestions to improve the translation of basic science findings to the clinic. Several of these workshops have indicated the need for multicenter approaches in preclinical studies so that the efficacy of interventions can be tested across the platforms. 14 –16
Accordingly, our groups have formed a consortium to investigate the preclinical efficacy of the mitochondria-targeting peptide, Bendavia. Bendavia (SS-31; MTP-131) is a tetrapeptide that has shown cytoprotective efficacy in a number of models (reviewed in 17 ). Our groups previously found that postischemic administration of Bendavia reduced several indices of reperfusion injury across experimental models, 18 and Bendavia is now in clinical trials for acute coronary syndromes. 19 Administration of Bendavia before the onset of reperfusion reduced the extent of myocardial infarction and coronary no-reflow in intact animal models and reduced cell death in isolated primary myocytes exposed to hypoxia/reoxygenation. The mechanism of protection involved reducing the extent of ROS-dependent cell death.
Although these results are promising, several issues regarding Bendavia’s mechanism of action remain to be resolved. First, the timing of Bendavia administration that is required for cardioprotection is not clear. As an adjuvant therapy, the effective treatment window must be clearly defined to maximize benefits to patients, especially since the onset and etiology of myocardial injury vary as a function of time. 20,21 Incomplete understanding of the timing of myocyte death during/after ischemic injury remains a major hurdle for effective treatments and is an aspect of cardioprotection that has been identified by experts in the field necessitating further study. 14,16 Second, the protective mechanisms for Bendavia is still not fully known. The magnitude of protection provided by Bendavia is consistent with direct blockers of the mitochondrial permeability transition pore (PTP), 18,22 –24 but whether Bendavia directly blocks PTP opening warrants in-depth investigation. Further, although Bendavia clearly reduces intracellular ROS levels in tissues exposed to oxidative challenge (reviewed in 17 ), whether this is due to a ROS-scavenging effect or lowered ROS production is not known. This is an important issue to address regarding the mechanism of action, as previously tested ROS scavengers have shown equivocal results in clinical trials. 25,26
The present study represents a collaboration spanning 4 distinct research centers. Several experimental models of acute myocardial infarction were utilized to test the hypothesis that Bendavia’s effective treatment window is occurring early in the reperfusion period. The capacity of Bendavia to alter mitochondrial respiration, directly block the PTP, and scavenge ROS was also tested and compared to several positive controls.
Methods
All animal experiments were conducted with prior approval from the Institutional Animal Care and Use Committee at each institution (Good Samaritan Hospital, University of Southern California, QTest Labs, East Carolina University, and the University of Missouri at Columbia). Rabbit studies were conducted at the Heart Institute at the Good Samaritan Hospital, University of Southern California and at QTest Labs in Columbus, Ohio. Guinea pig studies were done at East Carolina University, and studies using mouse mitochondria were done at the University of Missouri at Columbia. The animals used in these studies were maintained in accordance with the policies and guidelines of the Position of the American Heart Association on research animal use (American Heart Association, 1985) and the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources, National Research Council (Department of Health, Education and Welfare Publication No. 85-230).
Rabbit Ischemia/Reperfusion and Coronary No-Reflow
Rabbit ischemia/reperfusion experiments were conducted in 2 study arms: (1) administration of Bendavia during the ischemic period (studies conducted at QTest Labs) and (2) administration of Bendavia after the onset of reperfusion (studies conducted at Good Samaritan Hospital).
For the administration of Bendavia during the ischemic period (Study Arm 1), anesthesia was induced intramuscularly with ketamine (35 mg/kg) and xylazine (5 mg/kg) in male New Zealand White rabbits. A catheter was placed in the ear vein, and anesthesia was maintained by continuous infusion of propofol (approximately 60 mg/kg/h). A solid-state catheter was inserted into the aortic root to measure heart rate and blood pressure at baseline and at 10 minutes of coronary artery occlusion (CAO). An additional catheter was placed into an ear vein to administer drug treatment. Body temperature was monitored and maintained within the physiological range using temperature-controlled water-filled blankets and lamps. The thorax was opened and the heart suspended in a pericardial cradle. Sutures were placed around the left circumflex artery at a point between its origin and the base of the heart and around the left anterior descending artery or one of its branches in order to produce a risk zone comprising 50% to 60% of the left ventricle (LV). The ends of the suture were threaded through a piece of tubing, forming a snare that was tightened to occlude the arteries. Following a stabilization period (∼30 minutes), the rabbits were subjected to 30 minutes of CAO followed by 3 hours of reperfusion. At 10 minutes after the onset of ischemia, rabbits received an infusion of Bendavia (0.05 mg/kg/h) or a similar volume of vehicle. Treatment was maintained for 60 minutes (n = 8) or for 180 minutes (n = 7). A separate group of rabbits received (prior to reperfusion) 25 mg/kg/h of the PTP blocker cyclosporine A (CsA; n = 6), following a treatment protocol previously shown to be cardioprotective in this model. 23,24,27 At the end of the reperfusion period, the ischemic risk area was assessed by retightening the coronary artery sutures and injecting Evans blue dye intravenously. The heart was removed and sliced, and the slices were incubated in triphenyltetrazolium chloride to delineate the areas of necrosis in the heart slices. Each slice was measured with a micrometer, photographed, and scanned. Areas of the risk region and areas of necrosis were measured using Image J and expressed as a percentage of the total LV area in each slice. The sum of the areas of the slices was used to quantify the risk region and the necrotic region in each heart. Risk and necrotic areas were expressed as a percentage of the area of the LV below the occlusion site. Infarct size was expressed as a percentage of the risk region.
For the administration of Bendavia after reperfusion (Study Arm 2), the methods used for the rabbit model of acute myocardial infarction in the Kloner laboratory have been described previously. 18,28 Briefly, ketamine (75 mg/kg) plus xylazine (10 mg/kg) anesthetized, open-chest male New Zealand White rabbits were used. Fluid-filled catheters were inserted into a carotid artery to measure hemodynamics and into the jugular veins to administer supplemental anesthesia (pentobarbital, ∼50 mg/kg/h) and to infuse the drug treatment. Body temperatures were monitored and maintained at approximately 38°C using a heating pad. The circumflex coronary artery or one of its major branches was isolated with a suture in order to produce a risk zone of approximately 30% of the LV. The rabbits were subjected to 30 minutes of CAO followed by 3 hours of reperfusion. After initiation of reperfusion, the rabbits were randomized to one of the 2 groups: group 1 received 0.05 mg/kg/h of Bendavia, by intravenous infusion starting at 10 minutes after coronary artery reperfusion and continuing throughout reperfusion; group 2 received an equivalent volume of saline starting at the same time point and continuing throughout reperfusion. The total volume infused was <6 mL. Heart rate and blood pressure were measured throughout the protocol.
At the end of the reperfusion period, 1 mL/kg of a 4% solution of thioflavin S was injected into the heart via a catheter placed into the left atrial appendage in order to define the region of no-reflow. Thioflavin S, a fluorescent green-yellow dye, stains endothelium, serves as a marker of perfusion, and is used as a standard marker for identifying zones of no-reflow. Viewed under ultraviolet light, regions of the heart that are perfused fluoresce brightly, while the no-reflow zone appears as a nonfluorescent, dark area. The coronary artery was then reoccluded, and the ischemic risk region was delineated with 4 mL of a 50% solution of Unisperse blue dye (Ciba-Geigy, Hawthorne, New York) injected into the left atrium. The KCl (12 mEq) was then injected into the left atrium, and the heart was excised, sliced, and photographed. The heart slices were then incubated in triphenyltetrazolium chloride to delineate areas of necrosis. Measurements of risk zone, no-reflow zone, and infarct size were calculated as described previously. 18,28 A total of 20 rabbits were randomized to the study. One rabbit randomized to the vehicle group was excluded: the epicardial surface of the heart in this rabbit displayed pink blushing (rather than cyanosis) when examined at 10 minutes after CAO. The clamp occluding the artery had in all probability loosened allowing reperfusion. This heart had nearly no necrosis (<2% of the LV) when examined at the end of the protocol. Final data are reported for 10 rabbits in the Bendavia group and 9 rabbits in the vehicle group.
Guinea Pig Ischemia/Reperfusion
Isolated guinea pig hearts were exposed to ischemia/reperfusion using protocols well established by the Brown Laboratory. 29 –31 Male guinea pigs (n = 13) were anesthetized using a pentobarbital cocktail (35 mg/kg; intraperitoneal injection), and hearts were excised after the absence of animal reflexes. Hearts were retrograde perfused with modified Krebs-Henseleit buffer on a Langendorff apparatus (perfusion pressure 75 mm Hg) and instrumented for the simultaneous measurement of left ventricular function, volume-conducted electrocardiogram, and coronary flow. After a 20-minute stabilization period, hearts were exposed to 20 minutes of global ischemia followed by 2 hours of reperfusion. Hearts were randomly assigned to receive either no drug (control group, n = 7) or Bendavia (1 nmol/L, n = 6) beginning at the onset of reperfusion and continuing for the duration of the reperfusion period. At the end of reperfusion, hearts were dissected and stained for infarction using tetrazolium salt staining.
Mitochondrial PTP Assays
Mouse liver mitochondria were isolated and assayed for PTP opening under energized and de-energized conditions using Dr Baines’ established protocols. 32 Briefly, 0.25 mg/mL mitochondria were placed in swelling buffer containing 120 mmol/L KCl, 10 mmol/L Tris/HCl, and 5 mmol/L KH2PO4 (pH 7.4), with (energized) or without (de-energized) 5 succinate (separate pilot experiments with glutamate/malate yielded similar results). Mitochondrial PTP opening was observed by a decrease in absorbance at 520 nm after the addition of 25 µmol/L (energized conditions) or 250 μmol/L (de-energized conditions) CaCl2. The effects of Bendavia were compared to several positive controls known to influence PTP opening. Prior to calcium pulses, mitochondria were treated with 100 μmol/L Bendavia, 1 mmol/L of the radical scavenger mercaptopropionylglycine (MPG), 10 μmol/L of the superoxide dismutase (SOD) mimetic manganese(III) tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP), or 10 μmol/L of the PTP blocker CsA. Absorbance data were collected every 10 seconds, normalized to baseline absorbance (absorbance change from 520 nm; ▵A520), and data are expressed as area above the curve. To confirm that PTP opening was evoked by the calcium pulse and not by the experimental reagents, separate experiments were conducted using a water vehicle control instead of CaCl2. There was no discernible effect of any of the treatments on PTP opening in the absence of calcium (data not shown).
Isolated Mitochondria Experiments
In separate studies, rat heart mitochondria were isolated and rates of respiration and H2O2 production were assessed using our established protocols. 22,33 Mitochondria of 200 μg were suspended in assay buffer containing 110 mmol/L K-MES, 35 mmol/L KCl, 1 mmol/L ethylene glycol tetraacetic acid, 5 mmol/L K2HPO4, 3 mmol/L MgCl2, 5 mg/mL BSA, and 10 mmol/L succinate (pH 7.4). State II (no adenosine diphosphate [ADP]) and state III (2.5 mmol/L ADP) respiration (normalized to protein content) were assayed in a Oxygraph O2K high resolution respirometer (Oroboros Insruments, Austria). Rates of respiration were assessed with substrate only (reverse electron flow) or with 2 μmol/L rotenone to block complex I (promoting forward electron flow). Mitochondrial H2O2 production was determined in assay buffer (no rotenone) containing 10 μmol/L Amplex Ultra Red (Invitrogen, Carlsbad, CA), 1 U/mL horseradish peroxidase, and 10 mmol/L succinate. H2O2 fluorescence was converted to H2O2 concentration using a standard curve. Rates of respiration and H2O2 emission were tested in the presence or absence of 1 nmol/L Bendavia or 400 U/mL catalase.
In Vitro ROS Scavenging Experiments
The ROS-scavenging capacity of Bendavia was determined with novel static and dynamic biochemical systems coupled with fluorometric measurements to measure ROS scavenging in cell-free systems. These enzymatic systems generate H2O2 and/or superoxide anion as reaction products. 34,35 Because the overwhelming majority of ROS during conditions of oxidative stress are likely nonradical oxidants, 36 the effect of Bendavia on scavenging H2O2 in cell-free systems was examined first. For static H2O2 scavenging studies, 2 mL of potassium phosphate reaction buffer was placed into 12-well plates, and 100 μmol/L H2O2 was added for 10 minutes. Each well contained either Bendavia (final concentrations of 1 nmol/L, 1 μmol/L, and 1 mmol/L) or the positive control H2O2-scavenger catalase (100 U/mL final concentration). The H2O2 levels were detected using 10 μmol/L Amplex Ultra Red (Invitrogen) and 1 U/mL horseradish peroxidase (485 nm excitation/590 nm emission wavelengths). Raw fluorescence intensities were converted to nmol/L of H2O2 using a standard curve.
The H2O2-scavenging capacity of Bendavia was also assessed using a glucose–glucose oxidase reaction system that dynamically generates H2O2. Glucose of 10 mmol/L was dissolved in reaction buffer, and 100 mU/mL of glucose oxidase were added to the wells to initiate the ROS-generating reaction. Each well contained either Bendavia (concentrations of 1 nmol/L, 1 μmol/L, and 1 mmol/L) or 100 U/mL catalase as a positive control. The reaction was allowed to run for 30 minutes, and at minutes 10, 20, and 30, samples were taken from the wells and incubated with AmplexRed/horseradish peroxidase. In separate experiments, inclusion of the SOD mimetic 2,2,6,6-tetramethylpyperidine-1-oxyl (1 mmol/L final concentration) or SOD (25 U/mL final concentration) in the reaction had no effect on the generation of H2O2, confirming that the dominant ROS produced by this reaction couple is H2O2. 34,35
Finally, the ability of Bendavia to scavenge superoxide anion was determined using a xanthine/xanthine oxidase system. Superoxide is an early product of the incomplete reduction of oxygen to water, and although it is likely short lived in the living systems, 36 it is believed to be a major contributor to reperfusion injury. 10 This reaction generates both superoxide and H2O2, 37 and we isolated the superoxide component by scavenging H2O2 throughout with catalase. For dynamic superoxide scavenging studies, 100 mU/mL of xanthine oxidase were added to potassium phosphate reaction buffer containing 50 U/mL catalase and 5 μmol/L of the superoxide sensor mitoSOX. Each well contained Bendavia at concentrations of 0 mmol/L (control), 1 nmol/L, 1 μmol/L, and 1 mmol/L, or 50 U/mL of the positive control superoxide dismutase. After a 6-minute period to obtain background fluorescence, 100 μmol/L of xanthine was added to initiate the reaction for 10 minutes, with the average peak fluorescence taken over the last 4 minutes. MitoSOX fluorescence was obtained by determining the peak fluorescence minus background, and data are presented as arbitrary fluorescence units.
Statistical Analysis
In studies with the 2 groups (Rabbit Study Arm 2 and guinea pig infarct size study), infarct sizes, areas at risk, and areas of no-reflow were compared using Student t test. In rabbit studies where Bendavia duration was altered (Rabbit Study Arm 1, data in Figure 1), as well as isolated mitochondria studies, data were analyzed with an analysis of variance (ANOVA), followed by Tukey’s multiple comparison post hoc test. Changes in hemodynamic variables over time were analyzed by ANOVA (repeated measures). Analysis of covariance was used to test for a group effect on the regression model of the no-reflow zone versus the risk zone. Data are expressed as mean ± standard error of the mean throughout the manuscript, and significance was declared when P < .05.
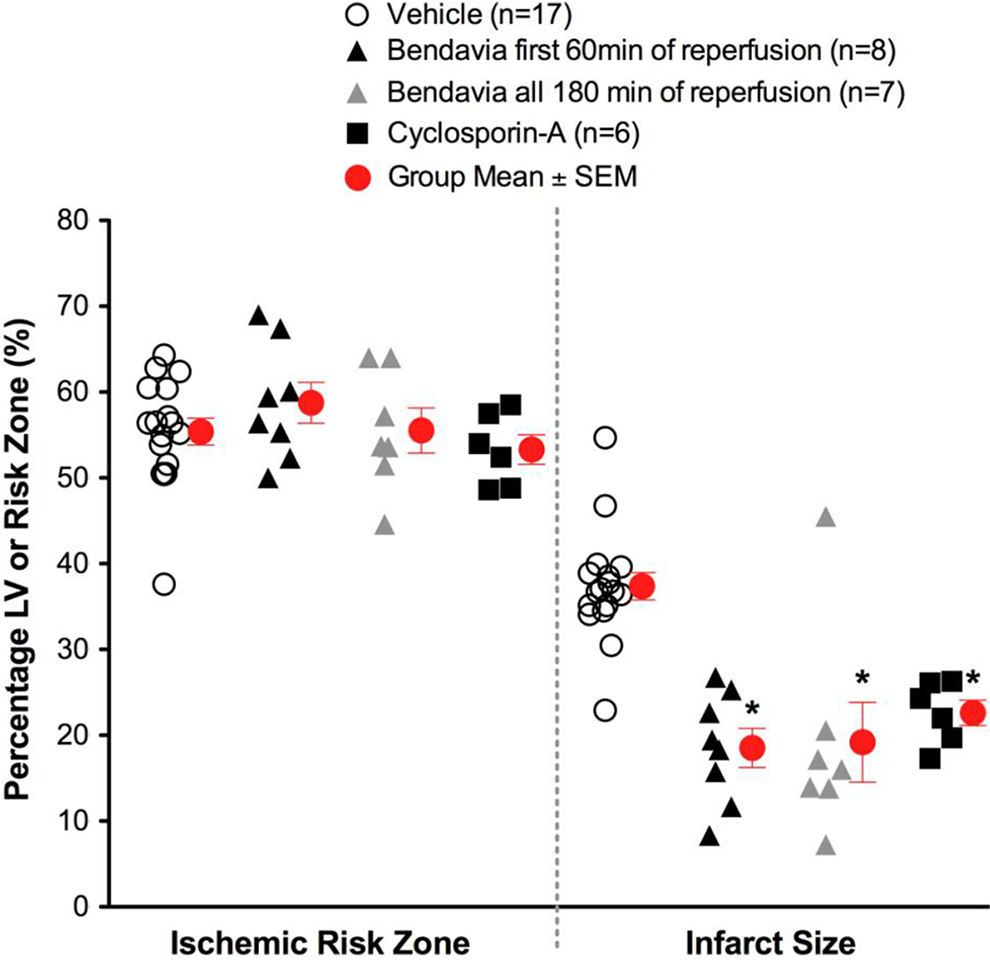
Scatter plots for ischemic risk zone (expressed as a % of the LV) and infarct size (expressed as a % of the risk zone) from rabbits exposed to in vivo ischemia/reperfusion (30 min/3 h, respectively). Bendavia infusion (0.05 mg/kg/h) via a marginal ear vein began 20 minutes prior to reperfusion and continued for either the first hour of reperfusion or all 3 hours of reperfusion. *P < .05 compared to vehicle (ANOVA). ANOVA indicates analysis of variance; LV, left ventricle.
Results
Rabbit Ischemia/Reperfusion
There were no differences in the size of the zones at risk among experimental groups. Postischemic administration of Bendavia significantly reduced infarct size in rabbits (Figure 1). Bendavia treatment protected the heart equally well whether Bendavia was administered for the entire 3-hour reperfusion period or just for the first hour of reperfusion (P < .05 for both treatments versus control). Bendavia was equally effective as the PTP blocker, CsA.
When Bendavia treatment was administered 10 minutes after reperfusion, cardioprotection was no longer observed. Infarct size was not different between placebo-treated and Bendavia-treated rabbits (Figure 2). Further, the no-reflow defect expressed either as a fraction of the risk zone or as a fraction of the necrotic zone was of similar size between the groups. There was no beneficial effect on the reduction of anatomic no-reflow when Bendavia treatment began 10 minutes after the onset of reperfusion (Figure 3).

Scatter plots for ischemic risk zone (expressed as a % of the LV) and infarct size (expressed as a % of the risk zone) from rabbits exposed to in vivo ischemia/reperfusion (30 min/3 h, respectively). Bendavia infusion (0.05 mg/kg/h) began 10 minutes after the onset of reperfusion. There were no differences in infarct size or area at risk between control and Bendavia-treated groups. LV indicates left ventricle.

Anatomic no-reflow in hearts given 0.05 mg/kg/h Bendavia beginning 10 minutes after the onset of reperfusion. A, No-reflow, expressed as either a percentage of the risk zone or a percentage of the necrotic zone, was no different between control and Bendavia-treated rabbits (P > .05 for both comparisons). B, Relationship between no-reflow zone and the ischemic risk zone. There was no significant group effect when Bendavia administration began 10 minutes after the onset of reperfusion (P > .05, ANCOVA). ANCOVA indicates analysis of covariance.
Bendavia treatment had no effect on heart rate or mean arterial blood pressure in either of the rabbit protocols. Similarly, there were no significant group differences in systolic or diastolic blood pressure (data not shown).
Guinea Pig Ischemia/Reperfusion
Administration of Bendavia at the onset of reperfusion significantly reduced the extent of infarction (P < .05; Figure 4). There were no differences in the extent of left ventricular developed pressure recovery, ventricular arrhythmia, rates of contraction or relaxation, or coronary flow in any of the treatment groups (data not shown). The extent of protection in these studies is similar to the cardioprotection we previously observed in guinea pig hearts, 18 with one key difference. The anesthetic used in these studies was pentobarbital, whereas before we used a ketamine/xylazine cocktail. Given that the anesthetic regimen can significantly influence infarct size in cardioprotection studies, 31,38 –40 the consistency across these studies with Bendavia treatment is encouraging.
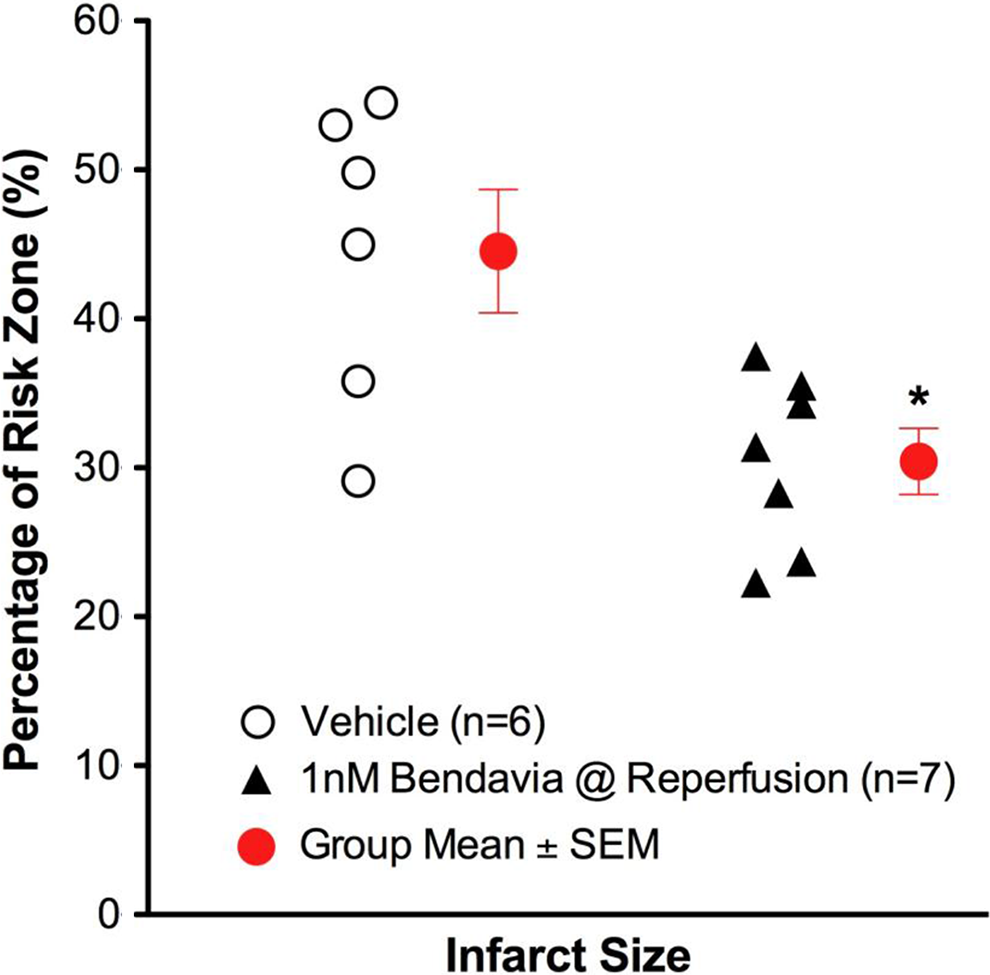
Infarct sizes in isolated guinea pig hearts exposed to 20-minute global ischemia and 2 hours of reperfusion. Bendavia treatment (1 nmol/L, dissolved in Krebs-Henseleit buffer) began at the onset of reperfusion and continued throughout the protocol. Since this is a global ischemia model, the risk zone is by definition 100% of the LV. *P < .05 versus control (Student t test). LV indicates left ventricle.
Isolated Mitochondria Experiments
Results on the effect of Bendavia on PTP opening in energized and de-energized mitochondria are presented in Figure 5A and 5B, respectively. The PTP opening was evoked by calcium pulses and is reflected by a decrease in absorbance at 520 nm. Regardless of the energetic status of the mitochondria, Bendavia had no effect on PTP opening, in clear contrast to the radical scavenger MPG, the manganese superoxide dismutase mimetic MnTBAP, and the direct PTP blocker CsA. The MnTBAP delayed PTP opening in energized but not de-energized mitochondria.
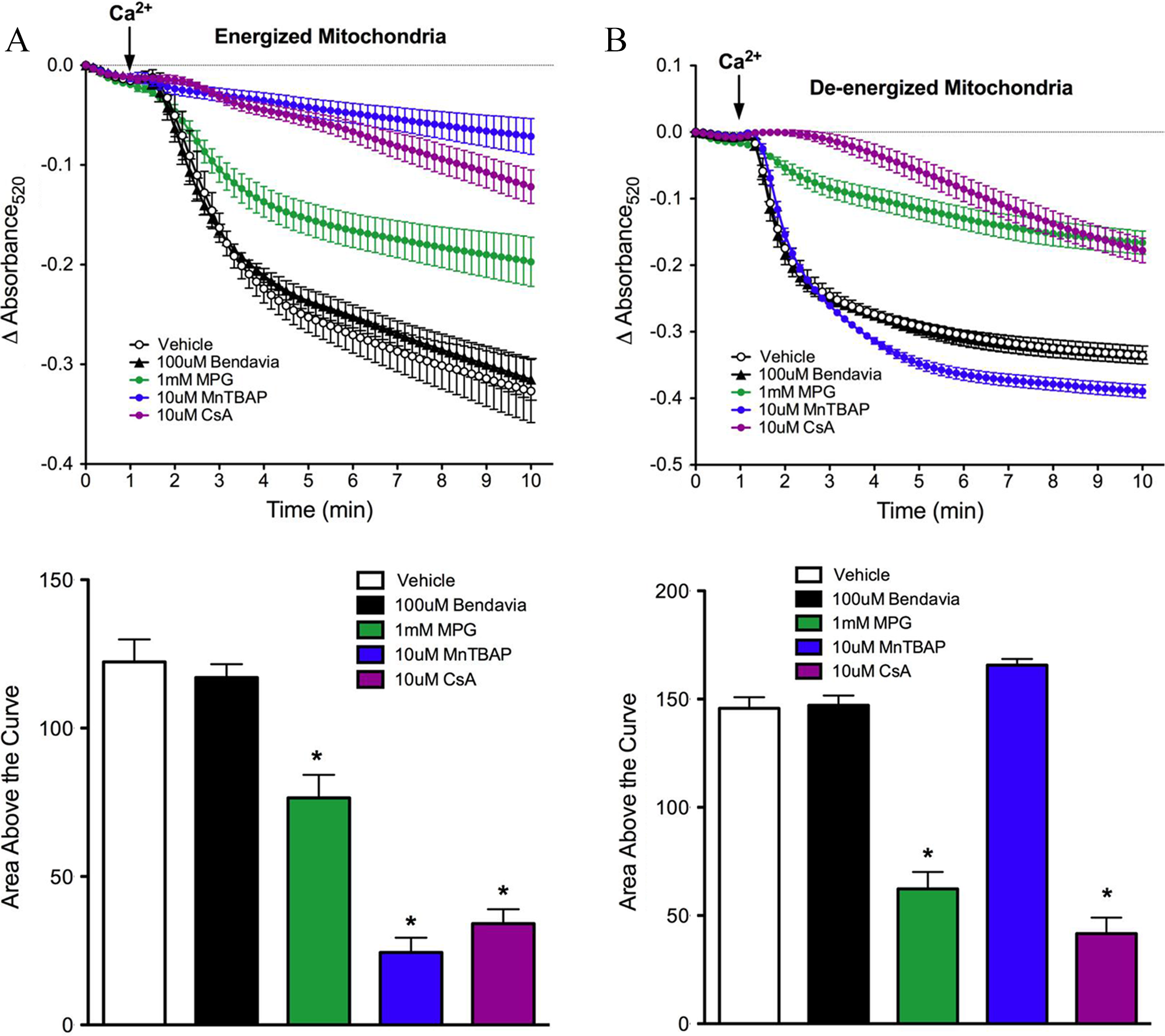
Lack of influence of Bendavia on PTP opening in isolated mitochondria subjected to a calcium pulse. A, Mitochondria were energized with glutamate/malate. The positive controls MPG (a radical scavenger), MnTBAP (a manganese superoxide dismutase mimetic), and CsA (a direct PTP blocker) were utilized for comparison with Bendavia. B, Experiments were repeated under de-energized conditions (no glutamate/malate). The PTP opening is depicted by mitochondrial swelling, which is reflected by a decrease in the absorbance of light at 520 nm. *P < .05 (ANOVA). ANOVA indicates analysis of variance; PTP, permeability transition pore; CsA, cyclosporine A; MPG, mercaptopropionylglycine; MnTBAP, manganese(III) tetrakis (4-benzoic acid) porphyrin chloride.
Mitochondrial respiration and H2O2 emission data are presented in Figure 6. Rates of state 2 and 3 respiration in heart mitochondria were not altered in the presence of Bendavia (Figure 6A) whether mitochondria were respiring in the presence of rotenone (forward electron flow) or not (reverse electron flow). Mitochondrial H2O2 emission was significantly reduced with both Bendavia and catalase (Figure 6B).
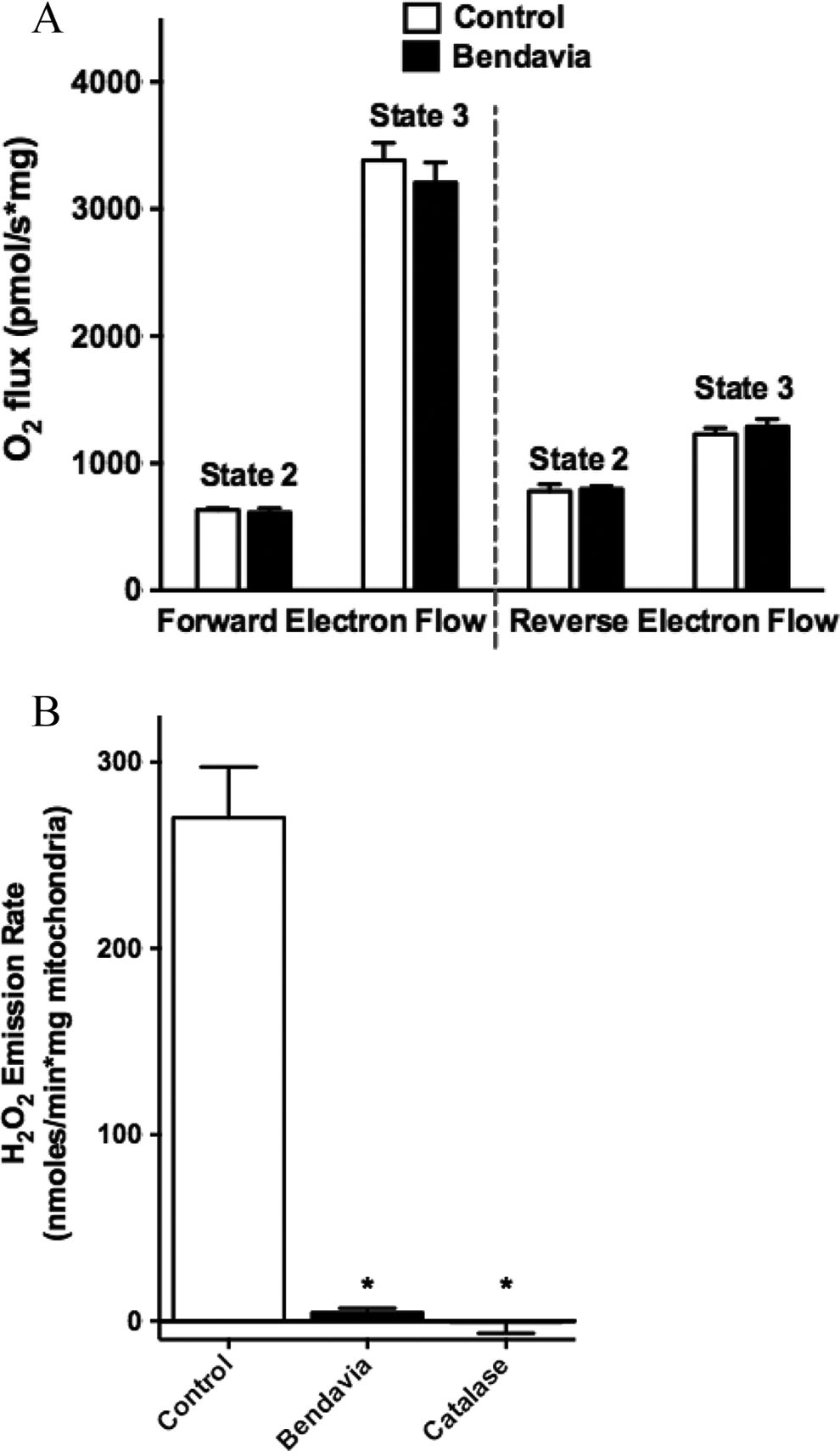
Effects of Bendavia on heart mitochondrial respiration and H2O2 emission. A, State 2 and 3 respiration in the presence (forward electron flow) or absence (reverse electron flow) of rotenone. B, H2O2 emission in mitochondria was significantly decreased with Bendavia or catalase. *P < .05 (ANOVA). ANOVA indicates analysis of variance.
In Vitro ROS Scavenging Capacity
Our studies examining the scavenging capacity of Bendavia are presented in Figure 7. The protective effects of Bendavia have been attributed to mitochondrial targeted ROS scavenging by the peptide. 17,33 To directly test this hypothesis, ROS production was measured in cell-free ROS-generating systems in the presence or absence of Bendavia. In both a static H2O2 incubation and a dynamic H2O2-generating system, Bendavia concentrations ranging from 1 nmol/L to 1 mmol/L had no effect on H2O2 levels, in clear contrast to the H2O2 reductions evoked by the positive control catalase (Figure 7A-C). Likewise, Bendavia over the same concentration range failed to alter the rate of superoxide generation in a cell-free superoxide-generating system, again in direct contrast to a positive control (superoxide dismutase; Figure 7D).

Lack of ROS-scavenging capacity of Bendavia in in vitro, cell-free systems. The ability of Bendavia to scavenge ROS (specifically H2O2) was tested with either a static H2O2 incubation (A), or a dynamic glucose–glucose oxidase reaction, which generates H2O2 as a reaction product (B). The net amount of H2O2 produced under each condition in B was calculated after 30 minutes of the glucose–glucose oxidase protocol and are presented in (C). The H2O2 levels were assessed by Amplex Red fluorescence. Bendavia did not influence H2O2 levels across a wide range of concentrations tested, in clear contrast to the positive control catalase. D, Additionally, the superoxide (O2 • −) scavenging capacity of Bendavia was tested using a xanthine–xanthine oxidase reaction. Bendavia also showed no observable scavenging capacity for O2 • −, in contrast to the positive control SOD. *P < .05 (ANOVA). ANOVA indicates analysis of variance; ROS, reactive oxygen species; SOD, superoxide dismutase.
Discussion
In this study, our groups tested the hypothesis that Bendavia, a novel mitochondria-targeting peptide, would reduce reperfusion injury in a specific time window. To the best of our knowledge, several aspects of this study represent novel findings. First, we show for the first time that administration of Bendavia is equally effective when given for either 1 hour or 3 hours of reperfusion. Second, treatment with Bendavia beginning just 10 minutes prior to the reperfusion period had absolutely no effect on the extent of myocardial infarction or the development of coronary no-reflow, indicating that the effective window for treatment occurs early in reperfusion. Third, Bendavia had no direct effect on blocking the PTP in either energized or de-energized mitochondria. Fourth, the protection elicited by Bendavia is independent of a direct ROS-scavenging mechanism. Taken together, this study enhances the understanding of how and when this cell-permeable, mitochondria-targeting peptide protects heart tissue from reperfusion injury.
Early Reperfusion: A Case for Targeting Mitochondria
Although prompt reperfusion remains the most effective therapy for acute myocardial infarction, reperfusion injury is a significant problem and can influence both short- and long-term survival after an infarction. 41 Reperfusion injury is multifactorial at the cellular level and varies as a function of time. Accordingly, therapies designed to reduce the burden of reperfusion injury must be given at the proper time to be effective. In general, early reperfusion is characterized by a burst in cellular (mitochondrial) ROS, calcium overload, and a restoration of intracellular pH, all of which promote the opening of mitochondrial pores (such as the PTP, reviewed in 21,42,43 ). In the later phases of reperfusion injury, apoptotic cell death predominates, and the infiltration of inflammatory cells begins to remodel the scar in the weeks (and perhaps months) after the insult.
Bursts of reactive oxygen injury were first described in the setting of acute myocardial infarction in the late 1980s. Bolli et al and Garlick et al used free radical spin traps testing the coronary effluent and observed a drastic (and transient) increase in ROS within the first 20 minutes of reperfusion, peaking within the first 5 minutes. 1,2,44 Subsequent studies have confirmed that mitochondria are a significant source of these ROS bursts, with the majority of ROS likely existing as nonradical oxidants (since free radicals are notoriously short-lived and rapidly dismutated to nonradical oxidant species). 36 Early attempts to reduce the mitochondrial ROS bursts included superoxide mimetics and glutathione precursors such as N-acetylcysteine. Although there were early promising results in animal models, 45 –47 these approaches are plagued with problems including permeability issues and side effects secondary to very high (millimolar) concentrations of the compounds. 48 –52 More recent strategies have employed antioxidants conjugated to mitochondria-targeting lipophilic cations (reviewed in 53 ). These approaches appear promising but require polarized mitochondria for effective targeting, which means that the compound may only be helpful to mitochondria that are adequately recovering from ischemia. Furthermore, as these compounds may depolarize mitochondria in high doses, these approaches may be self-limiting, with a narrow therapeutic window. 17
Cardioprotection With the Mitochondria-Targeting Peptide Bendavia
The reduction in myocardial infarct size observed herein is consistent with previous studies using Bendavia (SS-31; MTP-131). 18,22,54 Central to the development of Bendavia into a clinical therapy is identification of the mechanism of action. A wide variety of models have previously suggested that Bendavia reduces intracellular ROS levels, most commonly assessed by fluorescence monitoring of H2O2 with fluorophores (summarized in 17,55,56 ). Our data in isolated heart mitochondria (Figure 6) also show a clear effect in keeping ROS emission low and are consistent with previous studies using mouse liver mitochondria. 57 These studies are promising, yet the findings mostly represent lowered ROS levels. As ROS levels represent a balance between production and scavenging, previous studies have not typically differentiated between improved ROS-scavenging versus lower overall ROS production with Bendavia. Herein, a novel series of in vitro, cell-free reaction systems reveal that Bendavia does not appear to function as a direct ROS scavenger. These findings suggest that Bendavia is targeting mitochondria and reducing ROS production by a mechanism distinct from electron scavenging, most likely through an as yet undefined interaction with the electron transport system. Very recent studies suggest that Bendavia may be targeting the mitochondrial phospholipid cardiolipin. 58 The activity of each of the mitochondrial proteins associated with electron transport depends on cardiolipin, 59 and Bendavia’s protective effect may involve optimizing cardiolipin microdomains to reduce electron leak from the inner membrane (recently reviewed in 60 ).
Our results in this study are consistent with the idea that strategies aimed at reducing the ROS burden will be most effective early in reperfusion. In a recent study, Kloner and colleagues showed that ROS-dependent cell death in primary cardiac myocytes exposed to hypoxia/reoxygenation was significantly blunted with Bendavia, resulting in substantially higher cell survival during early reperfusion (cell death in Bendavia-treated cells was similar to untreated cells during the hypoxia period). 18 The data presented herein underscore the importance of therapeutic timing in early reperfusion. In intact rabbits exposed to ischemia/reperfusion, our groups previously observed that Bendavia was effective in reducing infarction and the extent of coronary no-reflow if started minutes prior to reperfusion 18 but not if started 10 minutes after reperfusion (current study). These findings are consistent with other models of cardioprotection such as activation of the reperfusion injury signaling kinase pathway 61 and “ischemic postconditioning,” 62 which also lose their effectiveness if not begun immediately at reperfusion. Although we cannot completely rule out damage prevented during ischemia that is attenuated with Bendavia, we previously found that Bendavia was equally effective if given 10 minutes after ischemia or 1 minute prior to reperfusion. 18 These data suggest that the early reperfusion window appears to be the crucial therapeutic window for cardioprotection with Bendavia.
Cardioprotection From Bendavia: Indirectly Keeping Mitochondrial Pores Closed
During this early reperfusion window, mitochondria often become overwhelmed by ROS bursts and display collapses/oscillations in mitochondrial membrane potential (▵Ψm). The loss of ▵Ψm hampers the ability of mitochondria to maintain adenosine triphosphate production and can lead to the onset of infarction, pump dysfunction, and arrhythmia (recently reviewed in 7 ). Attempts to maintain ▵Ψm often focus on keeping specific mitochondrial pores/channels closed, with the mitochondrial PTP being the most common target examined (including in recent human clinical trials 63 ). Consistent with the idea of maintaining bioenergetic integrity, Bendavia leads to sustainment of ▵Ψm in cardiomyocytes during ischemia/reperfusion, 18 suggesting that Bendavia might act as a direct PTP blocker. Additional support for this notion came from studies where Bendavia was quantitatively as cardioprotective as strategies (such as CsA or NIM811) that directly block the mitochondrial PTP (Figure 1 and 18,22 ).
The findings of the present study significantly extend the knowledge about Bendavia and the PTP, showing that the ability of Bendavia to sustain ▵Ψm is probably indirect. Since mitochondria in the reperfused heart display very heterogeneous energetics (ie, mitochondria can be polarized, depolarized, or oscillating between energetic states 64,65 ), it is imperative to investigate the effects of mitochondria-targeting compounds across mitochondrial energetic states. In this study, Bendavia had no direct PTP-blocking effects in isolated mitochondria regardless of the energetic status, in clear contrast to CsA and well-known ROS scavengers. Since ROS production is known to promote PTP opening, 5,6 it is tempting to speculate that Bendavia promotes tissue salvage/viability by lowering ROS production and indirectly keeping the PTP closed. We can also rule out mitochondrial uncoupling as a protective mechanism with Bendavia, as there was no effect of Bendavia on the rate of respiration regardless of energetic state (Figure 6).
In general, the magnitude of cardioprotection provided by Bendavia is proportional to the extent of injury. For example, in studies where the zone-at-risk comprises >50% of the LV, the magnitude of protection by Bendavia is highest. The rabbit data in Figure 1 (zone at risk was >50% of the LV) and the guinea pig data in both Figure 5 and Kloner et al 18 (where 100% of the LV was ischemic) showed a 40% to 50% reduction in infarct size with Bendavia. In previous sheep and rabbit models 18 where the risk zone comprised a smaller portion of the LV (25% to 30% of the LV), the extent of protection with Bendavia was smaller, averaging a 10% to 20% reduction. These findings are consistent with a recently published clinical trial, 63 where cardioprotection with cyclosporine was the most prominent in patients with the largest areas at risk (also noted by Downey and Cohen 12 ).
Several limitations to our study/interpretations are worth noting. First, we found that Bendavia did not block the PTP in isolated mitochondria, but these studies were performed in liver mitochondria. Although the data are consistent with our previous work in heart mitochondria, 22 we did not investigate PTP opening in heart mitochondria herein. Second, we have shown that ▵Ψm is better maintained in cells during hypoxia/reoxygenation with Bendavia. 18 We cannot rule out that Bendavia may have PTP-blocking effects in intact cells by interacting with endogenous regulators of PTP in isolated cells (which may be washed out in isolated mitochondrial experiments).
Another limitation in this study is that we did not determine the efficacy of Bendavia alongside current standard-of-care therapies for patients with acute coronary syndromes. Specifically, very recent studies suggest that platelet inhibitors, which patients would normally receive, can themselves confer cardioprotection by activating protective signaling cascades. 66 Future studies determining whether there are additive effects of Bendavia alongside platelet inhibitors will be an important advancement. Furthermore, as the rabbit experiments in Figures 1 and 2 were conducted at different academic institutions, the area at risk varied between the models. This is likely due to different locations of coronary occlusion and/or different strains of rabbit used across groups. Finally, the long-term efficacy of Bendavia in the context of ischemia/reperfusion injury must be determined. Given that chronic Bendavia treatment shows efficacy in preserving cardiac function in models of heart failure, 67,68 the long-term benefit of postischemic Bendavia treatment is of significant interest.
In conclusion, our groups have taken a consortium approach to understand how and when this mitochondria-targeting peptide protects the heart. This study provides novel evidence that Bendavia remains an attractive candidate for the reduction of cardiac reperfusion injury if administered early in reperfusion, and that the mechanism of action appears to be distinct from direct PTP block, mitochondrial uncoupling, or ROS scavenging.
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: DA Brown, PD Neufer, and RA Kloner have served as consultants for Stealth Peptides.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was supported in part by the American Heart Association (Pre-Doctoral Fellowship to CR Frasier and DA Brown), NIH grant DK096906 (to PD Neufer), and Stealth Peptides.