
Abstract
The objective of this study is to examine whether carbon monoxide-releasing molecules (CORMs) can decrease the generation of excessive reactive oxygen species (ROS) in cardiac mitochondria, thereby protecting against postresuscitation myocardial injury and cardiac mitochondrial dysfunction after resuscitation in a rat model of ventricular fibrillation (VF), and further investigated the underlying mechanism. Rats suffered 8 minutes of untreated VF and resuscitation and were randomized into the control group with vehicle infusion and the CORM group with CO-releasing molecule 2 (CORM2) treatment. Animals in the Sham group were instrumented without induced VF and resuscitation. Effects of CORM2 on cardiac function, myocardial oxidative stress, cardiac mitochondrial function, and mitochondrial ROS generation were assessed. Moreover, to further evaluate the direct effect of CORM2 on cardiac mitochondria isolated from resuscitated rats, we measured mitochondrial function and ROS generation when isolated cardiac mitochondria were directly incubated with different concentrations of (CORM2). Compared with the Sham group, the control and CORM groups demonstrated impaired cardiac function, increased myocardial injury, and aggravated mitochondrial damage. CORM2 improved cardiac performance and attenuated myocardial damage and oxidative stress in resuscitated rats. Additionally, animals with CORM2 treatment showed the decreased generation of cardiac mitochondrial ROS, alleviated mitochondrial injury, and preserved mitochondrial function and complex activities when compared with the control group. In isolated cardiac mitochondria incubated with CORM2, low concentrations of CORM2 (20 μmol/L) mildly uncoupled mitochondrial respiration, leading to reduced mitochondrial ROS production. In contrast, high concentrations of CORM2 (60 μmol/L) resulted in the reverse effect presumably due to its excessive uncoupling action. These findings suggest that CORM2 attenuates oxidative stress of the heart and improves cardiac function after resuscitation. The mechanism was probably that CO, the product of CORM2, reduces the production of cardiac mitochondrial ROS and thereby attenuates mitochondrial injury and dysfunction during the postresuscitation period, due to the transient uncoupling of mitochondrial respiration.
Introduction
With the development of cardiopulmonary resuscitation (CPR) science and techniques, the rate of restoration of spontaneous circulation (ROSC) in cardiac arrest has substantially increased, but the mortality rate after ROSC is still high. 1,2 Postcardiac arrest myocardial dysfunction is one of the most common causes of early death in patients following resuscitation. 3 A significant body of experimental and clinical evidence indicates that the overproduction of reactive oxygen species (ROS) plays an important role in cardiac injury and myocardial dysfunction during the postresuscitation period. 4 –7 This view was further substantiated on discovering the beneficial effects of antioxidants on hearts subjected to ischemia/reperfusion damage. 5 The enhanced generation of ROS is a result of several enzymatic reactions, including xanthine oxidase in endothelial cells and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in inflammatory cells. 8,9 Moreover, the mitochondrial electron transport chain is thought to be the major source of ROS during reperfusion, especially in hearts containing a great quantity of mitochondria. 10 Excessive electron leakage from the mitochondrial respiratory chain leads to the increased formation of mitochondrial ROS following ROSC, such as superoxide anion, hydrogen peroxide, and so on. 11 Meanwhile, mitochondria are also the major attack targets of ROS and the core determinants of myocardial dysfunction after cardiac arrest. The impairment of mitochondrial function leads to a decrease in energy production, induction of myocardial apoptosis, and further overproduction of ROS. 12 In some in vitro experiments, the transient blockade of mitochondrial electron transport, resulting in a decrease in electron leakage, can substantially attenuate the bust generation of ROS and thus protect the mitochondria and the cardiac function in hearts with ischemia/reperfusion injury. 13,14 It is conceivable that the damage to the cardiac mitochondria and myocardial function may be mitigated by the reduction in ROS generation at the site of the cardiac mitochondria in postcardiac arrest myocardial injury as a kind of global ischemia/reperfusion damage.
Carbon monoxide (CO) is known as a toxic gas that interferes with O2 delivery because of its high affinity for hemoglobin. In mammalian cells, CO as a product of heme oxygenases has various biological functions such as anti-inflammation, vasorelaxation, among others to protect cells against certain forms of injury and disease. 15 It is corroborated that low concentrations of CO can promote protection against ischemia/reperfusion injury, although the specific mechanisms are not well established. 16 Recently, CO-releasing molecules (CORMs) have emerged as better compounds for delivering CO due to their predictable release kinetics and the control of a stabilized concentration of CO. 17 The CORMs represent a group of CO carriers that have been used in a series of studies. 18 Berne et al found that CO-releasing molecule 2 (CORM2) decreased oxidative stress to protect against ischemia/reperfusion injury in isolated rat hearts via antioxidant properties. 19 Additionally, Lo Iacono et al suggested that low concentrations of CORMs, as observed in an in vitro study, may reduce the reverse electron flow and the ROS production of mitochondria in situations with excessive ROS produced. 20 Thus, CORMs may be a potential pharmacological remedy to cardiac damage after resuscitation for protecting cardiac mitochondria and myocardial function by decreasing excessive ROS generation of cardiac mitochondria. Nevertheless, although the effect of CORMs on attenuating mitochondrial ROS production in in vitro experiments has been found, it is unknown whether such a paradigm can be extended to postresuscitation myocardial dysfunction in clinics. Additional work, especially in resuscitation models different from previous in vitro experiments, may be important to further confirm and elucidate this protective mechanism of CORMs. For this purpose, the present study examined whether CORM2, a type of CORMs, could decrease excessive ROS generation of cardiac mitochondria, thereby protecting postresuscitation myocardial function and cardiac mitochondria after ROSC in a rat model of ventricular fibrillation (VF) and further investigated the underlying mechanism.
Materials and Methods
Animal Preparation and the Establishment of an Animal CPR Model
Forty-five male Sprague-Dawley rats (380-420 g) were cared in accordance with the Chinese Guidance Suggestions for the Care and Use of Laboratory Animals and with the approval of the Animal Care and Use Committee of Sun Yat-sen University. Animals were anesthetized by an injection of 45 mg/kg intraperitoneal (ip) pentobarbital sodium and required to maintain adequate anesthesia during the whole experimental procedure by additional doses of 10 mg/kg ip pentobarbital sodium. The trachea was orally intubated. The left femoral artery was cannulated with polyethylene (PE) 50 catheters into the aorta for monitoring arterial pressure. A PE-50 catheter was advanced from the right carotid artery into the left ventricle (LV) for hemodynamic monitoring. The right external jugular vein was cannulated by a 4F PE catheter and advanced into the right atrium for electrical induction of VF and infusion of CORM2 after ROSC. Cardiac rhythm was monitored and recorded by electrocardiogram. Hemodynamic data were recorded by a WinDaq data acquisition system (DataQ, Akron, Ohio). Body temperature was maintained at 36.8°C ± 0.2°C.
After instrumentation, the animals were mechanically ventilated with a tidal volume of 0.6 mL/100 g, fraction of inspired oxygen (Fi
Chemicals
CORM2 and inactive CORM2 (iCORM2) were purchased from Sigma Chemicals (St Louis, Missouri). CORM2 was dissolved in normal saline containing 1% dimethyl sulfoxide (DMSO). The CORM2 fresh solution was made just before use. iCORM2 with the same basic structure as CORM2, which cannot liberate CO, was diluted in the same condition as CORM2.
Experimental Procedure
Animals were randomly assigned into CORM group (CORM, n = 16), control group (control n = 16), and Sham group (Sham, n = 8). In the CORM group, animals were administrated CORM2 (4 mg/kg, dissolved in 1% DMSO and diluted in normal saline) at the onset of ROSC (within 15 minutes) after 8 minutes of cardiac arrest via the right external jugular vein. In the control group, animals were administrated an equivalent content of vehicle (4 mg/kg iCORM2, diluted in the same condition as CORM2) at the same time point. Animals in the Sham group were instrumented without induced VF and CPR. Animals in both the CORM group and the control group were again randomly divided into 2 subgroups based on the time of ROSC (1 and 3 hours after ROSC; Figure 1, protocol 1). To investigate the direct effect of CORM2 on isolated cardiac mitochondria, another 5 resuscitated animals after 8 minutes of cardiac arrest were killed immediately after ROSC. The cardiac mitochondria were isolated and randomly divided into the control group with vehicle and the CORM group with different concentrations of CORM2 (Figure 1, protocol 2).
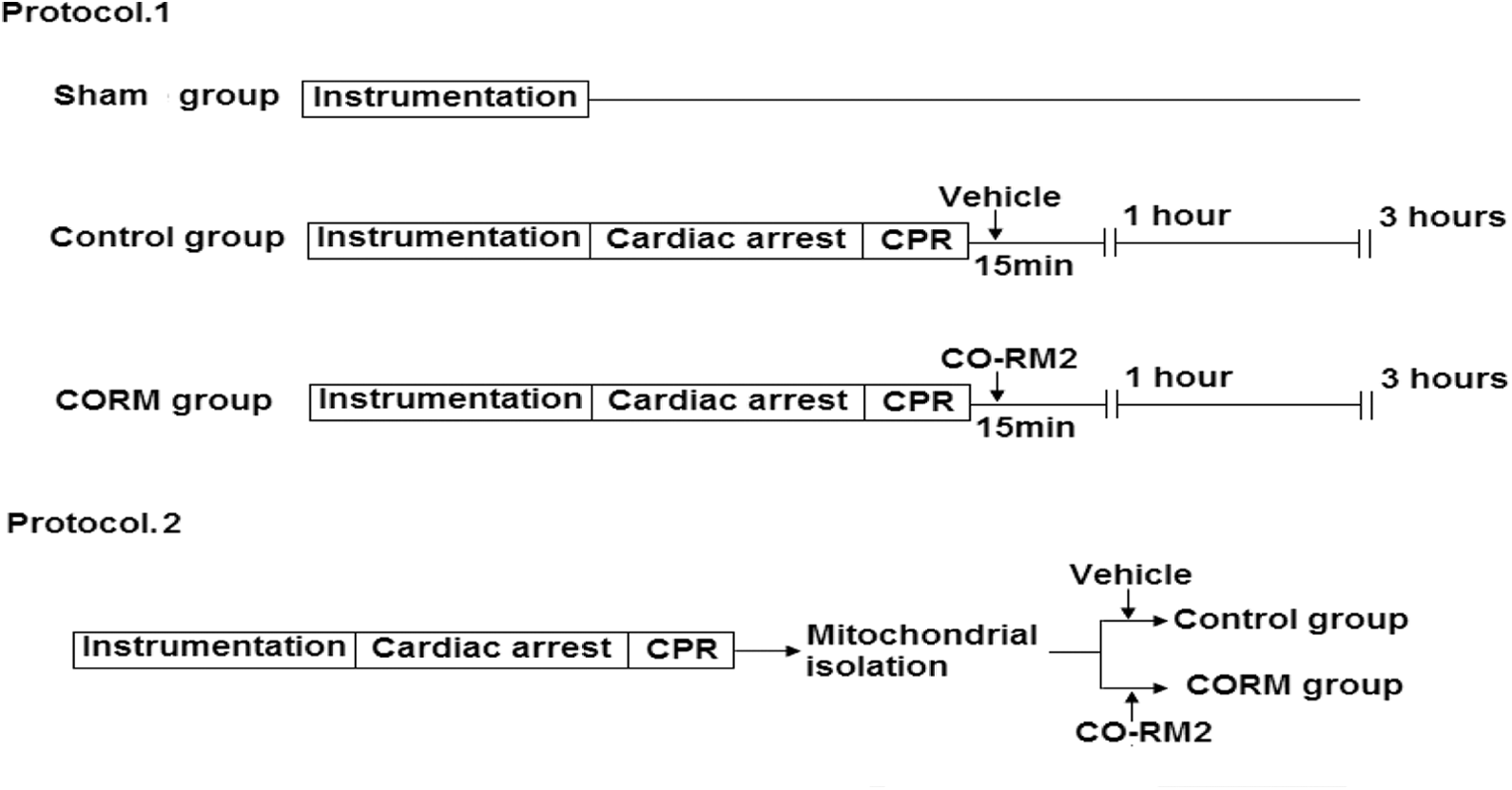
Experimental procedure. Protocol 1 shows the design of the in vivo experiment performed in cardiopulmonary resuscitation (CPR) rats. Protocol 2 shows the design of the ex vivo experiment performed in cardiac mitochondria isolated from the hearts of resuscitated rats. In protocol, CORM2, CORM2 dissolved in 1% DMSO and diluted in normal saline; vehicle, iCORM2 diluted in the same condition as CORM2. CORM2 indicates carbon monoxide-releasing molecule 2; iCORM2 inactive carbon monoxide-releasing molecule 2.
Detection of Serum Creatine Kinase MB Levels and Carboxyhemoglobin Levels in Blood Samples
Creatine kinase MB (CK-MB) levels in serum were assayed using a Rat CK-MB Elisa Kit (CUSABIO, China) according to the manufacture’s instructions and expressed as ng/L. Carboxyhemoglobin (COHb) level in blood samples was detected using an OSM3 Hemoximeter (Radiometer, Denmark).
Detection of Myocardial ROS Production
Myocardial ROS production was measured using the Tissue ROS Classical Assay Kit (GENMED, Boston, Massachusetts) that utilized 2′,7′-dichlorofluorescein diacetate as the oxidative fluorescent probe. After the fresh LVs were isolated, the levels of myocardial ROS were measured following the manufacturer’s instructions within 1 hour.
Determination of Malondialdehyde in Myocardial Tissue
Malondialdehyde (MDA) as the production of lipid peroxidation was measured using a tissue MDA determination kit (GENMED, Boston, Massachusetts). Malondialdehyde forms a conjugate with thiobarbituric acid (TBA; MDA-TBA2) resulting in the red production (532 nm absorbance). 21 The MDA concentration was rectified for protein concentration and expressed as mmol/mg protein.
Isolation of Cardiac Mitochondria
Hearts were harvested after ROSC. Mitochondria were isolated by differential centrifugation of heart homogenates and resuspended in mitochondrial storage buffer following the manufacturer’s instructions of the Mitochondria extraction Kit (GENMED, Boston, Massachusetts). Mitochondrial protein concentration, expressed as mg mitochondrial protein/L, was measured by the Lowry method using bovine serum albumin as standard. Fresh mitochondria were used for detecting respiration and ROS. Mitochondria, stored at −80°C, were used for other analyses.
Determination of Mitochondrial ROS Production
Mitochondrial ROS production was measured using the mitochondrial ROS testing kit (GENMED, Boston, Massachusetts). Cardiac mitochondria were loaded with the working reagent containing the fluorescent probe 6-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) at 37°C for 15 minutes. The fluorescence was measured at 530 nm following excitation at 490 nm using Fluorescence spectrophotometric enzyme mark instrument (SpectraMax M5, San Francisco, California). The results were corrected for the mitochondrial protein concentration in the samples.
Determination of Cardiolipin in Mitochondrial Membranes
Cardiolipin, a phospholipid localized almost exclusively within the mitochondrial inner membrane, is an early target of ROS attack. 22,23 The cardiolipin of mitochondria, which were not peroxided, were analyzed to evaluate the peroxidation damage of mitochondrial inner membrane. 24 The intact cardiolipin was measured using the mitochondria peroxidation/damage determination Kit (GENMED, Boston, Massachusetts), which used nonyl-acridine orange (NAO) as the main assay reagent. The results were rectified for the mitochondrial protein concentration.
Measurement of Mitochondrial Respiration
The oxygen consumption of fresh mitochondria was measured using a Clark-type oxygen electrode at 25°C. Mitochondria were added to the respiration buffer (GENMED, Boston, Massachusetts) with or without CORM2. Mitochondrial respiration (state 2) was initiated by the addition of 2 mmol/L pyruvate + 5 mmol/L malate. After 1 minute, state 3 respiration was induced by adding adenosine diphosphate (ADP; 0.5 mmol/L). On depletion of ADP, state 4 respiration was measured. Respiratory control ratio (RCR) was defined as the ratio of state 3 to state 4. The ADP to O ratio was calculated according to the total oxygen consumption during state 3 respiration.
Measurement of Respiratory Enzymes Activity
Complex I activity was assayed by monitoring the dynamic change in transmittance from oxidation of NADH to NAD+. Complex III activity was analyzed by ubiquinol-mediated ferricytochrome c reduction. The activities of complex I and complex III were measured using the animal mitochondrial respiratory enzymes complex I and complex III activities quantitative determination Kit (GENMED, Boston, Massachusetts). The mitochondria under test were prepared by sonicating under nitrogen gas. The activities of the respiratory enzymes were, respectively, expressed as nmol/NADH/min/mg protein and nmol/cytochrome C/min/mg protein.
Pathological Examination of Myocardial Tissue
Hearts were excised at 3 hours after ROSC. Left ventricular tissue sample were partially embedded in paraffin, cut into 6-μm thick sections, and stained with hematoxylin and eosin. In each microscopic field, myocytolysis, transverse contraction bands, and organization of myocardial fiber were investigated under an optical microscope by 1 experienced pathologists who was unaware of any experimental data.
Statistical Analysis
Statistical analysis was performed using SPSS 17.0 software (SPSS Inc, Chicago, Illinois). Data were presented as mean ± standard deviation. Statistical analyses were carried out with 1-way analysis of variance for comparisons of more than 2 groups. For 2-group comparison, an unpaired 2-sample t test was used. A value of P < .05 was considered statistically significant.
Results
Carbon Monoxide-Releasing Molecule 2 Improved MAP and Cardiac Performance in ROSC Rats
There was no significant difference in baseline characteristics and hemodynamic data in each group before inducing cardiac arrest (Table 1, all P > .05). The COHb levels in blood samples were slightly improved in the CORM groups (0.76% ± 0.14% at 1 hour after ROSC and 0.43% ± 0.12% at 3 hours after ROSC vs 0.35% ± 0.07% in the Sham group), but they were far below the toxic level. Heart rate did not differ significantly in all groups after resuscitation (Figure 2A, all P > .05). However, the values of MAP, dp/dt40, and −dp/dtmax were significantly decreased in both the control and CORM groups compared with the Sham group (Figure 2B–D, all P < .01). One hour after ROSC, the MAP and −dp/dtmax were statistically increased in the CORM group compared to the control group (Figure 2B and D, both P < .05), and dp/dt40 was also significantly improved (Figure 2C, P < .01). Moreover, CORM2 treatment improved the postresuscitation cardiac performance presented by MAP, dp/dt40, and −dp/dtmax at 3 hours after ROSC compared to the control group (Figure 2B–D, all P < .01).

Postresuscitation hemodynamic data in the study. A, Heart rate. B, Mean aortic pressure. C, dP/dt40. D, Maximal negative dP/dt. Values are mean ± standard deviation (SD). *P < .01 versus Sham group; # P < .01 versus control group; and † P < .05 versus control group.
Baseline Characteristics and Hemodynamic Data Before Inducing Cardiac Arrest.

Abbreviations: CORM, carbon monoxide-releasing molecule; MAP, mean aortic pressure.
Carbon Monoxide-Releasing Molecule 2 Attenuated Myocardial Injury and Oxidative Stress After ROSC
The CK-MB, an intracellular enzyme, was measured to assess the degree of cardiac injury. There was a significant increase in CK-MB in the control and CORM groups compared with the Sham group (Figure 3A, all P < .01). However, at 1 and 3 hours after ROSC, the levels of CK-MB were significantly reduced in the CORM groups as compared to the control groups (Figure 3A, P < .05 and P < .01, respectively). Pathological examination was also performed to evaluate the myocardial damage in ROSC rats. At 3 hours following ROSC, the control group showed myocytolysis and transverse contraction bands (Figure 4B1 and B2). The animals treated with CORM2 exhibited less severe myocytolysis and alleviation of disordered myocardial fibers (Figure 4C1 and C2). These findings suggest that CORM2 could mitigate myocardial injury after resuscitation. To evaluate myocardial oxidative stress, we detected both myocardial ROS production and MDA in myocardial tissue. The myocardial ROS production was increased in both the control and CORM groups over the Sham group but significantly reduced in the CORM groups compared to the control groups at 1 and 3 hours after ROSC (Figure 3B, all P < .01). Likewise, at both 1 and 3 hours after ROSC, MDA in myocardial tissue was significantly decreased in the CORM groups compared to the control groups (Figure 3C, both P < .01). Figure 3C also shows that the values of MDA in myocardial tissue were significantly higher in the control and CORM groups than the Sham group (both P < .01). These data indicate that CORM2 may protect the heart through ameliorating oxidative stress in ROSC rats.

The CK-MB levels and myocardial oxidative stress after restoration of spontaneous circulation (ROSC). A, Serum CK-MB levels. B, Myocardial reactive oxygen species (ROS) production. C, Myocardial malondialdehyde levels. Values are mean ± standard deviation (SD). *P < .01 versus Sham group; # P < .01 versus control group; and † P < .05 versus control group. CK-MB indicates creatine kinase MB.

Pathological examination of myocardial tissue at 3 hours after restoration of spontaneous circulation (ROSC). A1 and A2, Sham group; B1 and B2, control group; C1 and C2, carbon monoxide-releasing molecule (CORM) group; black arrow, myocytolysis; and red arrow, transverse contraction bands.
Carbon Monoxide-Releasing Molecule 2 Reduced the Generation of Cardiac Mitochondrial ROS and Attenuated the Injury of Cardiac Mitochondria in ROSC Rats
Mitochondrial ROS generation was detected when respiratory substrates were added to the isolated cardiac mitochondria. The generation of ROS was significantly elevated in the cardiac mitochondria from the control and CORM groups, compared with Sham group (Figure 5A, all P < .01). However, mitochondria in the CORM groups produced less amount of ROS than the control groups at 1 and 3 hours after ROSC (Figure 5A, both P < .01). Meanwhile, mitochondrial cardiolipin, a distinctive mitochondrial protein and the major attack target of ROS, was also tested to evaluate the degree of mitochondrial peroxidation damage. As seen in Figure 5B, the content of mitochondrial cardiolipin was significantly decreased in both the CORM and control groups compared with the Sham group (all P < .01). Mitochondria in the control groups have significantly less content of cardiolipin than the CORM groups at 1 and 3 hours after ROSC (Figure 5B, both P < .01). These findings show that CORM2 mitigates the oxidative injury of cardiac mitochondria in the early stage of postresuscitation. We also measured RCR to assess cardiac mitochondrial function. The RCR of cardiac mitochondria was significantly decreased in the resuscitated rats from the control and CORM groups compared with the rats without cardiac arrest and CPR in the Sham group (Figure 6A, all P < .01). The decline in mitochondrial RCR was due to the significantly decreased state 3 respiration (Figure 6B, all P < .01), and no significant difference was found in the state 4 respiration between each group (Figure 6C, all P > .05). Moreover, CORM2 treatment significantly ameliorated the decrease in state 3 respiration (Figure 6B, P < .01 and P < .05, respectively) and improved the mitochondrial RCR compared to the control groups at 1 and 3 hours after ROSC (Figure 6A, both P < .01). We next tested the activities of complex I and complex III, probably as the main sites of mitochondrial ROS production. 11 Figure 7 shows that cardiac mitochondria in the control and CORM groups represented a significant decrease in the complex I and complex III activity, compared with the Sham group (all P < .01). The CORM2 treatment significantly attenuated the decline in the activities of both these enzymes at 1 and 3 hours after ROSC (Figure 7, all P < .01 or P < .05). These results demonstrate that CORM2 treatment mitigates the impairment of respiratory enzymes and thus reduces mitochondrial ROS production, leading to the attenuation of mitochondrial damage and dysfunction after resuscitation.

Oxidative stress in cardiac mitochondria after restoration of spontaneous circulation (ROSC). A, Cardiac mitochondrial reactive oxygen species (ROS) production. B, Cardiac mitochondrial cardiolipin. Values are mean ± standard deviation (SD). *P < .01 versus Sham group; # P < .01 versus control group.
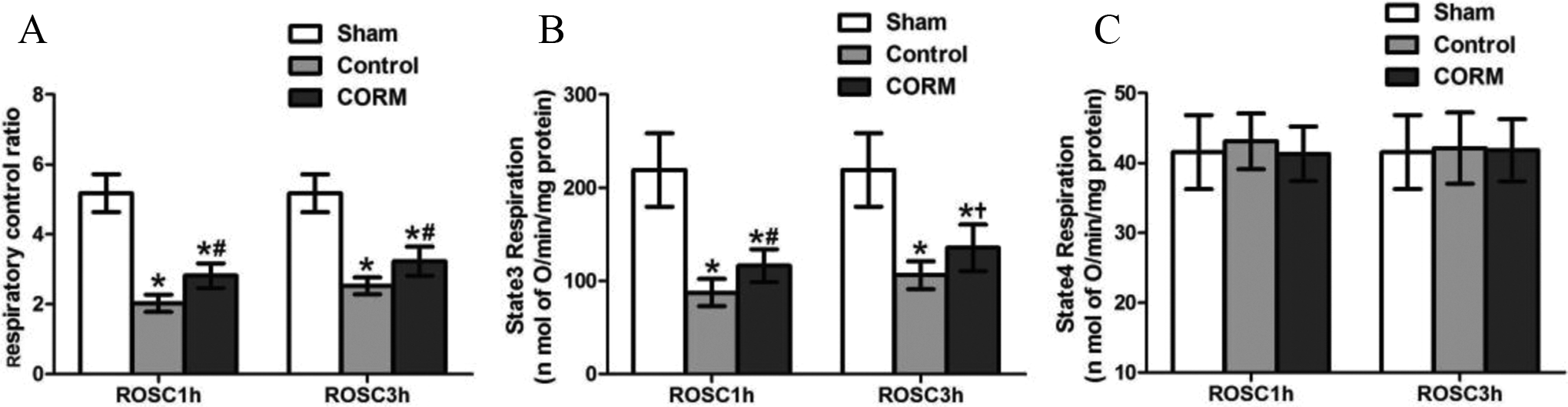
Mitochondrial respiration. A, Respiratory control ratio. B, State 3 respiration. C, State 4 respiration. Values are mean ± standard deviation (SD). *P < .01 versus Sham group; # P < .01 versus control group; and † P < .05 versus control group.
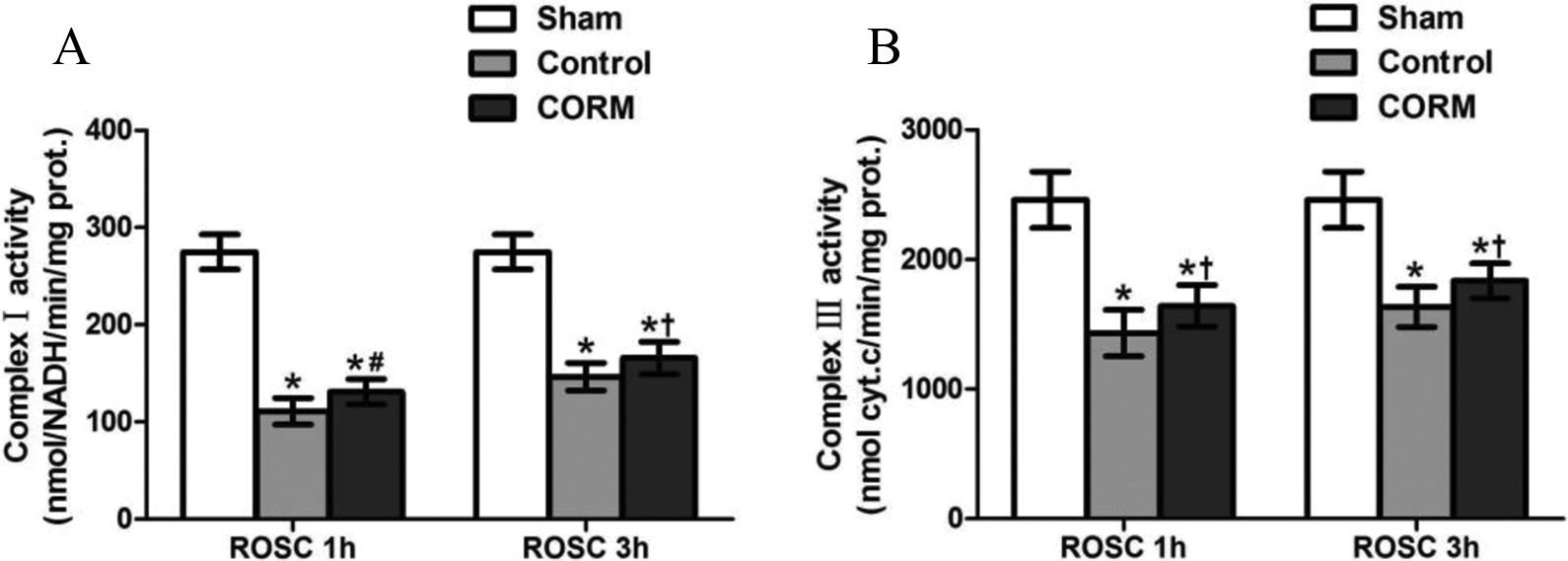
Activities of respiratory enzymes in cardiac mitochondria. A, Complex I activity. B, Complex III activity. Values are mean ± standard deviation (SD). *P < .01 versus Sham group; # P < .01 versus control group; and † P < .05 versus control group.
Low Concentrations of CORM2 Mildly Uncoupled Mitochondrial Respiration to Decrease Cardiac Mitochondrial ROS Production
To further evaluate the direct effect of CORM2 on cardiac mitochondria isolated from resuscitation rats, we measured the mitochondrial function and ROS generation when isolated mitochondria were directly incubated with different concentrations of CORM2. Meanwhile, we also detected mitochondrial ADP to O ratio to assess the tightness of the coupling between respiration and phyosphorylation. In the study, we observed that isolated cardiac mitochondria with 20 μmol/L CORM2 treatment produced significantly less ROS than the control group (Figure 8A, P < .01). In contrast, 60 μmol/L CORM2 treatment significantly increased mitochondrial ROS generation (Figure 8A, P < .01). Additionally, 60 μmol/L CORM2 decreased state 3 respiration (Figure 8E, P < .01), mitochondrial RCR, and ADP to O ratio (Figure 8B and C, both P < .01), but there was no statistical difference in the state 2 and state 4 respiration compared to the control group (Figure 8D and F, both P > .05). Cardiac mitochondria treated with 20 μmol/L CORM2 showed a slight decrease in RCR (Figure 8B, P > .05) and a significant decline in ADP to O ratio (Figure 8C, P < .05), with a statistical increase in state 2 respiration (Figure 8D, P < .01). There was no significant difference in the state 3 and state 4 respiration compared with the control group (Figure 8E and F, both P > .05). These findings indicate that low concentrations of CORM2 (20 μmol/L) mildly uncouple mitochondrial respiration probably due to the increase in state 2 respiration, leading to the reduction in electron leak and mitochondrial ROS formation in ROSC rats. On the contrary, high concentrations of CORM2 (60 μmol/L) result in a reverse effect presumably due to its excessive uncoupling action.

Respiratory activity and ROS production in isolated cardiac mitochondria incubated with carbon monoxide-releasing molecule 2 (CORM2). A, Mitochondrial ROS production; (B) respiratory control ratio; (C) adenosine diphosphate (ADP)/O ratio; (D) state 2 respiration; (E) state 3 respiration; and (F) state 4 respiration. Values are mean ± standard deviation (SD). # P < .01 versus control; † P < .05 versus control. ROS indicates reactive oxygen species.
Discussion
In the present study, we show that CORM2, a type of sustained CO donor, attenuates postresuscitation myocardial injury and protects mitochondrial function by decreasing abnormal ROS generation of cardiac mitochondria in ROSC rats. Several evidence in the study support this conclusion. First, CORM2 improved MAP and alleviated cardiac dysfunction, myocardial injury, and oxidative stress after ROSC. Second, excessive ROS generation of cardiac mitochondria was decreased during the postresuscitation stage in the CORM groups. Moreover, the decline in mitochondrial ROS production was evidently associated with the alleviation of mitochondrial injury and dysfunction. Third, in the injured cardiac mitochondria isolated from resuscitated rats, low concentrations of CORM2 mildly uncoupled mitochondrial respiration and dampened the electron leak, thus reducing the production of mitochondrial ROS. These findings indicate that CO, the product of CORM2, has a kind of antioxidant property, which is represented as decreasing mitochondrial ROS production by slightly uncoupling mitochondrial respiration, leading to the attenuation of cardiac injury in ROSC models. Several previous studies demonstrated the antioxidant effects of CO in ischemic–reperfusion models but focused on the improvement in antioxidant substances, such as the increase in superoxide dismutase 2, glutathione, and glutathione disulfide levels. 16,19,25 Moreover, these studies usually adopted an isolated ischemic–reperfusion heart model and some other in vitro experiments. There is a paucity of studies on CO or CORMs using animal CPR models as a global ischemic–reperfusion injury. So the origin of our study is to state that CORM2 attenuates myocardial damage and mitochondrial injury in animal CPR models by mitigating excessive ROS production of cardiac mitochondria and the mechanism may be that low concentrations of CORM2 mildly uncouple mitochondrial respiration in injured cardiac mitochondria.
Just like other previous in vitro studies, 19,26 we found that CORM2 attenuates myocardial injury and preserves cardiac function in well-established cardiac arrest rat models induced by VF. The substantial evidence lied in the decreased level of serum CK-MB, alleviated microscopic changes, and the improved cardiac index in the CORM groups compared with the control groups at 1 and 3 hours after ROSC. Furthermore, the results also showed that the ROS production and the MDA content of cardiac tissue were significantly cut down in the CORM groups, indicating CORM2 can mitigate the oxidative stress of heart during postresuscitation period. These findings suggested that the alleviation of myocardium damage and the preserved cardiac function after ROSC were in part due to the attenuation of oxidative stress by CORM2. This is consistent with previous evidences from some in vitro experiments. 19,27
Oxidative stress in ischemic–reperfusion injury is usually caused by excessive ROS generation overwhelming antioxidant defense system. Cardiac mitochondria are the major source of ROS in hearts subjected to ischemia-reperfusion damage. 11 Moreover, excessive ROS can lead to the damage of mitochondrial membrane and impaired mitochondrial function, which result in further mitochondrial ROS production, cardiac cell apoptosis, and decreased energy generation, forming a vicious cycle. 13 Mitochondrial injury and dysfunction are tightly associated with postresuscitation cardiac dysfunction, mainly due to cardiac mitochondria’s key roles on cell metabolism and apoptosis. 12 Hence, we further investigated the effect of CORM2 on mitochondrial ROS generation and mitochondrial function. First, we found CORM2 decreased ROS generation of cardiac mitochondria in the CORM groups compared with the control groups at 1 and 3 hours after ROSC. Additionally, we also determined the content of cardiolipin. Cardiolipin, an almost exclusive phospholipid within the mitochondrial inner membrane, was close to the site of ROS generation at mitochondrial respiratory chain and easily damaged by ROS. 24,28 Thus, mitochondrial cardiolipin was used to evaluate oxidative stress of mitochondria. In our study, NAO, a cardiolipin-specific assay reagent, was used to measure the content of mitochondrial cardiolipin. However, some reports demonstrated that NAO cannot be used to quantify cardioliplin in living cells since it does not bind exclusively to cardioliplin in vivo. 29,30 Jacobson et al supported this view but stated that isolated mitochondria, different from those in living cells, can be stably stained by NAO probably due to free of some cell-related factors affecting NAO loading and retention. 31 Hence, based on this observation, we decided to apply NAO in evaluating the cardioliplin content in isolated mitochondria in our study. In the current study, we found the content of cardiolipin was significantly improved in the CORM groups compared to the control groups at 1 and 3 hours after ROSC. This is consistent with the change in the mitochondrial ROS production in the CORM groups. These findings show that CORM2 mitigates oxidative stress of cardiac mitochondria after ROSC, presumably due to the decrease in mitochondrial ROS generation. Interestingly, we also observed the level of mitochondrial ROS was decreased at 3 hours after ROSC compared with 1 hour after ROSC in both the CORM and control groups. These results were further supported by the more content of cardiolipin at 3 hours after ROSC than 1 hour after ROSC. This suggests that oxidative stress may play an increasingly important role in the early stage of postresuscitation and thus early intervention may be more effective. Second, in our study, we detected mitochondrial RCR and the activities of complex I and complex III. In the postresuscitation stage, it was demonstrated by previous studies that mitochondrial oxidative phosphorylation, usually evaluated by RCR, was reduced. 32,33 Mitochondrial damage can lead to the decrease in ATP generation and thereby the electron leak at the sites of complex I and complex III to form excessive ROS. 11 These abnormal ROS further aggravate mitochondrial injury and dysfunction. Under serious oxidative stress, mitochondrial permeability transition pore could be abnormal opening, leading to cell apoptosis and death. 24 Consistent with the previous evidences, 21,22,34,35 we also found that the mitochondrial RCR and the activity of complex I and complex III were significantly reduced in cardiac mitochondria from both the control and CORM groups when compare to the sham group. In addition, we observed that the decrease in RCR and activities of complex I and complex III were significantly alleviated after the CORM2 treatment at 1 and 3 hours after ROSC, indicating CORM2 attenuates cardiac mitochondrial injury and partially preserves mitochondrial function. Previous studies demonstrated that the generation of mitochondrial ROS is positively related to the level of mitochondrial injury. 13,14 We speculate that the improved mitochondrial function by CORM2 occurs by attenuating the overproduction of mitochondrial ROS after ROSC. However, the effect of CO on mitochondrial RCR, which was used to evaluate the tightness of the coupling between respiration and phyosphorylation, is controversial. Sandouka et al found that CO decreased the respiratory control index in isolated mitochondria from kidney. 34 In contrast, Almeida et al reported that a brief exposition to CO improved the mitochondrial respiratory ratio in astrocytes isolated from cortex. 36 Moreover, previous studies suggested CO may have a 2-stage effect on mitochondrial cytochrome c oxidase, representing early effect as the decrease in COX activity and late effect as the increase in COX activity. 36,37 Lancel et al have also observed that low concentrations of CO could increase the mitochondrial RCR and high concentrations of CO have the reverse effect. 35 These controversial data may be due to different CO concentrations, different test time, and different modes. Accordingly, we further investigated the early effect of different concentrations of CO on isolated cardiac mitochondria from resuscitated rats. In our study, we found that high concentrations of CORM2 (60 μmol/L) led to the significantly decreased mitochondrial RCR and the increased generation of mitochondrial ROS compared with the control group. Interestingly, low concentrations of CORM2 (20 μmol/L) could also result in a litter decrease (P > .05) of mitochondrial RCR but significantly attenuating the ROS production of cardiac mitochondria when compared to the control group. Moreover, the ADP/O ratios were also significantly reduced, indicating an uncoupled mitochondrial respiration. In injured mitochondria with disrupted oxidative metabolism, mild uncoupling can reduce electron leak and ROS production. 38 These data show that low concentrations of CORM2 decrease the excessive ROS production of cardiac mitochondria in the early stage, possibly due to mildly uncoupling mitochondrial respiration. In contrast, excessive exposure to CORM2 exaggerates the injury of mitochondria, in part due to seriously suppressed mitochondria RCR and thereby the increase in mitochondrial ROS generation. These results were supported by Iacono et al study. 20 In isolated normal cardiac mitochondria, Iacono et al also found that the limited concentrations of CO could induce a mild uncoupling to alleviate excessive mitochondrial ROS production. 20 However, these findings may be a litter different with the previous results, where we observed CORM2 elevated mitochondria RCR with the attenuation of mitochondria ROS production at 1 and 3 hours after ROSC, compared with the ROSC rats without CORM2 treatment in our study. We speculate that the opposite is presumably due to the different period of CO exerting effects on injured cardiac mitochondria. In previous studies, it is reported that CO can reversibly bind to its molecular targets, highly depending on the system oxygen concentration. 26 Accordingly, we further speculate that CO may bind to the mitochondrial molecular targets to uncouple mitochondrial respiration in the early stage, and the uncoupling effect may be weakened following the restoration of oxygen. The early intervention of CO or CO donor can effectively alleviate mitochondrial ROS production, possibly leading to beneficial effects on the preservation of mitochondrial function and respiratory enzymes at 1 and 3 hours after ROSC. Although we furthermore found that CORM2 (20 μmol/L) significantly increased state 2 respiration and lightly decreased state 3 respiration in isolated cardiac mitochondria, indicating the uncoupling effect of CORM2, the specific sites of mitochondria. CORM2 makes effects on are uncertain and need to be further studied.
Our study has some limitations. First, we only observed the short-term effect of CORM2 on oxidative stress and cardiac function after ROSC. Although oxidative stress mainly occurs in the early reperfusion stage, postresuscitation cardiac dysfunction recovers after 24 hours and oxidative injury can last several days. 3 To further evaluate the long-term effect of CORM2, studying postresuscitation oxidative stress and cardiovascular improvement in long-term survival models is required. Second, we only focused on the effect of CORM2 on cardiac mitochondrial ROS production. The other ROS sources, such as NADPH oxidase, and antioxidants were not studied.
In conclusion, our study demonstrates that CORM2 attenuates the oxidative stress of the heart and improves the cardiac function after resuscitation. The mechanism was probably that CO, the product of CORM2, results in the decrease of cardiac mitochondrial ROS production and thereby attenuates mitochondrial injury and dysfunction during the postresuscitation period, in part due to transiently uncoupling mitochondrial respiratory in the early stage.
Footnotes
Author Contributions
Lan Yao contributed to conception or design, acquisition, analysis, or interpretation, drafted the article, critically revised the article, gave final approval, and agreed to be accountable for all aspects of work ensuring integrity and accuracy. Peng Wang and Zitong Huang contributed to conception or design, acquisition, analysis, or interpretation, critically revised the article, gave final approval, and agreed to be accountable for all aspects of work ensuring integrity and accuracy. Mingdi Chen, Yuanshan Liu, Lili Zhou, and Xiangshao Fang contributed to conception or design, acquisition, analysis, or interpretation, critically revised the article, and agreed to be accountable for all aspects of work ensuring integrity and accuracy. Lan Yao and Peng Wang contributed equally to this study and shared first authorship.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.