
Abstract
In the past 10 years, mortality from acute myocardial infarction has not decreased despite the widespread introduction of percutaneous coronary intervention. The reason for this situation is the absence in clinical practice of drugs capable of preventing reperfusion injury of the heart with high efficiency. In this regard, noteworthy natriuretic peptides (NPs) which have the infarct-limiting effect, prevent reperfusion cardiac injury, prevent adverse post-infarction remodeling of the heart. Atrial natriuretic peptide does not have the infarct-reducing effect in rats with alloxan-induced diabetes mellitus. NPs have the anti-apoptotic and anti-inflammatory effects. There is indirect evidence that NPs inhibit pyroptosis and autophagy. Published data indicate that NPs inhibit reactive oxygen species production in cardiomyocytes, aorta, heart, kidney and the endothelial cells. NPs can suppress aldosterone, angiotensin II, endothelin-1 synthesize and secretion. NPs inhibit the effects aldosterone, angiotensin II on the post-receptor level through intracellular signaling events. NPs activate guanylyl cyclase, protein kinase G and protein kinase A, and reduce phosphodiesterase 3 activity. NO-synthase and soluble guanylyl cyclase are involved in the cardioprotective effect of NPs. The cardioprotective effect of natriuretic peptides is mediated via activation of kinases (AMPK, PKC, PI3 K, ERK1/2, p70s6 k, Akt) and inhibition of glycogen synthase kinase 3β. The cardioprotective effect of NPs is mediated via sarcolemmal KATP channel and mitochondrial KATP channel opening. The cardioprotective effect of brain natriuretic peptide is mediated via MPT pore closing. The anti-fibrotic effect of NPs may be mediated through inhibition TGF-β1 expression. Natriuretic peptides can inhibit NF-κB activity and activate GATA. Hemeoxygenase-1 and peroxisome proliferator-activated receptor γ may be involved in the infarct-reducing effect of NPs. NPs exhibit the infarct-limiting effect in patients with acute myocardial infarction. NPs prevent post-infarction remodeling of the heart. To finally resolve the question of the feasibility of using NPs in AMI, a multicenter, randomized, blind, placebo-controlled study is needed to assess the effect of NPs on the mortality of patients after AMI.
Introduction
Despite the widespread use of coronary artery stenting in clinical practice, mortality from acute myocardial infarction is about 5% and has not decreased in recent years. 1 -3 The widely used percutaneous coronary intervention (PCI) allows for recanalization of the infarct-related coronary artery in 95% of cases. 4 With the use of PCI in clinical practice, reperfusion injuries of the heart play an increasing role, accounting for up to 50% of infarct size. 5 Hence, a search is underway for new pharmacological approaches to protect the heart from ischemia-reperfusion injuries: some of these approaches are based on the use of endogenous mechanisms to increase heart’s tolerance to ischemia and reperfusion. 6,7 Of particular interest in this regard are natriuretic peptides (NPs). The objective of this review is to summarize published data on the mechanisms of the cardioprotective effect of NPs, and to assess perspectives for the clinical use of NPs.
In 1981, the de Bold’s group found that rat atrial extracts had a diuretic effect and enhanced urinary sodium excretion. 8 Two years later they found that a 28-amino acid peptide had a natriuretic effect. 9 According to more recent data, it is composed of 29 amino acids. 10 In 1984, another group of researchers discovered that there are 2 atrial natriuretic peptides (ANPs): α-ANP and β-ANP. 11,12 In 1988, brain natriuretic peptide (BNP) was isolated from pig brain tissue and it was found that this molecule consists of 32 amino acids. 10,13 In 1990, a third type of natriuretic peptide: C-type natriuretic peptide (CNP), consisting of 22 amino acids, was isolated from pig’s brain (Figure 1). 14 It was later shown that ANP and BNP are synthesized in large quantities in myocardial tissue: ANP in the atrium and BNP in the ventricle and in the atrium. 15,16 Although ANP was found in both atrial and ventricular tissues, the ANP levels in the atria are 250–1,000 times higher than those in the ventricles. 16 The BNP level is higher in the atria, but since the weight of the atria is much less than the ventricles, the ventricular content of BNP is 30% higher than in the whole heart. 16 It has been demonstrated that human cardiac fibroblasts also express BNP. 17 It was found that CNP is synthesized not only in the brain, but also in peripheral organs and tissues. 15,16 ANP and BNP are mainly synthesized by atrial and ventricular cells, and enter the blood stream from there. 10 CNP is synthesized by endothelial cells, including endothelial cells of the coronary arteries, 18 and it is believed that the endothelium is the main source of circulating CNP. 19,20 NPs are sensitive to enzymatic hydrolysis. Hence, their half-lifes in plasma are short, for CNP 2.6 min, 21 for BNP 20 min, 22 and for ANP 2.4 ± 0.7 min. 23

Types of natriuretic peptides (NPs), receptors (NPRs) and their regulatory functions (Reg).
It is well known that NPs regulate renal function, affect nervous and endocrine signaling, energy metabolism, and the cardiovascular system, and play an important role in the regulation of blood pressure (BP) (Figure 1). 16,20,24 Natriuretic peptides are known as antihypertensive hormones. They have natriuretic and diuretic effects, cause vasorelaxation, and have anti-proliferative, anti-inflammatory and anti-hypertrophic effects (Figure 2). 24,25 NPs are autocrine and paracrine mediators which are released by cardiomyocytes, endothelial cells, and fibroblasts and regulate vital physiological functions in the cardiovascular system. 26 They control of inflammation, angiogenesis, smooth muscle and endothelial cell proliferation, atherosclerosis, cardiomyocyte contractility, hypertrophy, and fibrosis. 26 ANP causes vasodilation of the coronary arteries. 27 ANP. BNP, and DNP (a 38 amino acid peptide) stimulate NPR-A, whereas CNP exhibits selectivity to NPR-B. 10 ANP, BNP, CNP and DNP activate NPR-C. 10 The receptors for NPs are represented by 3 types: NPR-A, NPR-B, and NPR-C. NPR-A is a dimer that interacts with PNP, MNP, DNP and is guanylyl cyclase; NPR-B is a dimer that interacts only with CNP and is also guanylyl cyclase. 10 NPR-C is also a dimer, it interacts with all NPs. NPR-C is coupled to the Gi-protein, which allows it to inhibit adenylyl cyclase and, accordingly, reduce cAMP synthesis. 10 ANP, BNP, and DNP (a 38 amino acid peptide) stimulate preferentially NPR-A, and CNP exhibits selectivity for NPR-B. 10 ANP, BNP, CNP, and DNP activate NPR-C (Figure 3). 10 cANP (4-23) is the selective agonist NHR-C. 28 mRNA of all 3 receptors was found in the heart, the most pronounced expression was shown for NPR-C. 29 NPRs are expressed in the atria to a greater extent than in the ventricles. 10 The ventricular guanylyl cyclase activity and the cGMP levels were decreased by 96% and 87%, respectively, in Npr1-/- mice. 30 Endothelial-specific deletion of Nppc, which encodes CNP, leads to endothelial dysfunction, hypertension, atherosclerosis, and aneurysm. 31 Administration of NPR-C agonists promotes vasorelaxation of isolated arteries and a reduction in blood pressure (BP) in wild-type animals that is diminished in mice lacking NPR-C. 31 Intravital microscopy studies showed that CNP increases the diameter of arterioles and capillaries. 32 The vasodilatatory effect of CNP were preserved in mice with endothelial NPR-B deletion, but abolished in mice lacking NPR-B in microcirculatory SMCs and pericytes. 32 Endothelium-specific deletion of CNP (ecCNP-/-), resulted in impaired coronary responsiveness to endothelium-dependent- and flow-mediated-dilatation in mice. 33
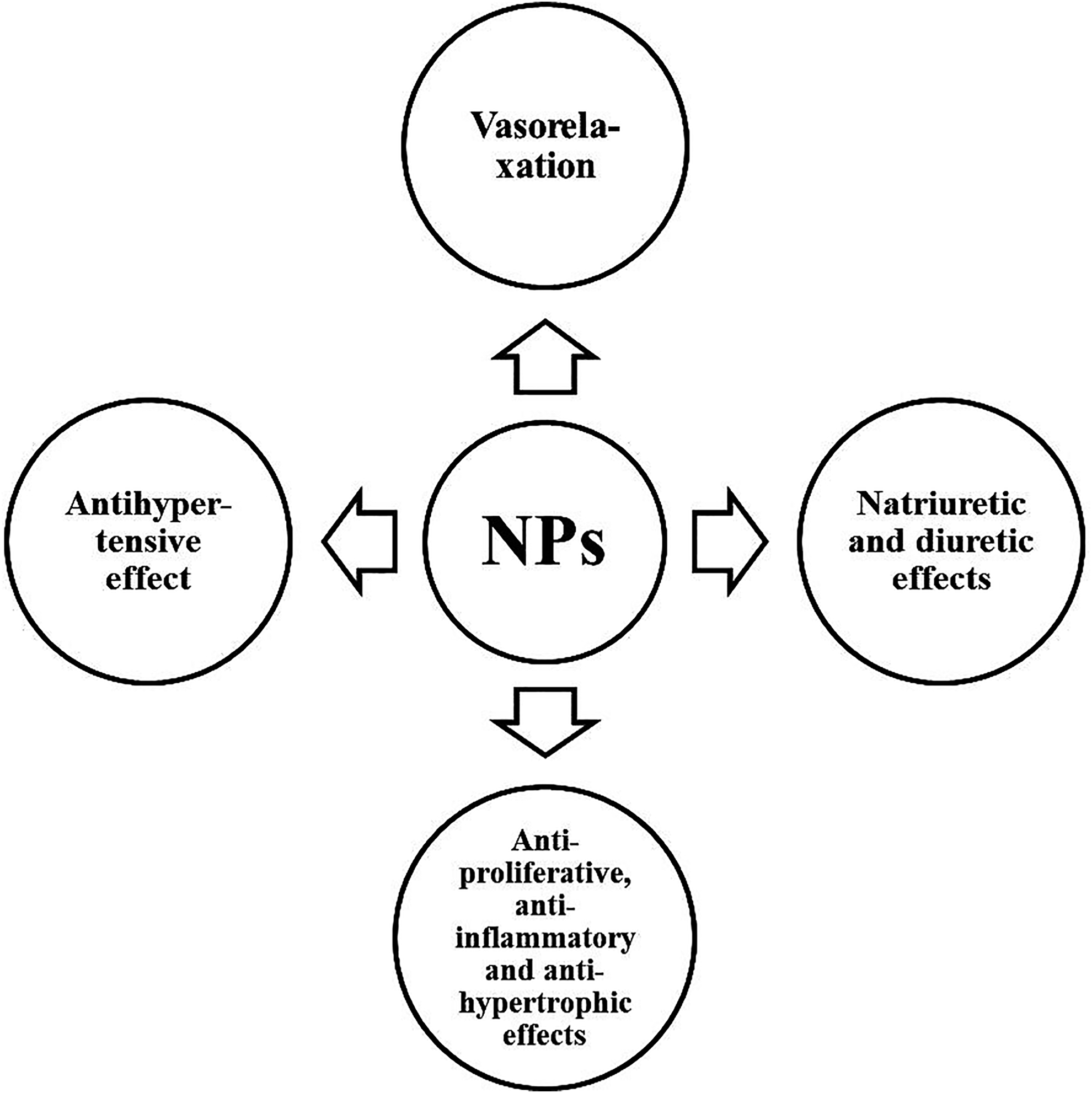
The main cardiovascular effects of natriuretic peptides (NPs).

Intracellular signaling pathway for the realization of the cardioprotective effect in activation of natriuretic receptors. AC, adenylate cyclase; Akt, protein kinase B; AMPK, cyclic adenosine monophosphateactivated kinase; ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide; cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; CNP, C-type natriuretic peptide; DNP, Dendroaspis natriuretic peptide; ERK1/2, extracellular regulated kinase; Gi, inhibitory G protein; GC, guanylate cyclase; GTP, guanosine triphosphate; HO1, hemeoxygenase-1; MEK(MAPK), mitogen-activated protein kinase; mitoKATP, mitochondrial ATP-dependent potassium channel; mPTP, mitochondrial permeability transition pore; NO, nitric oxide; NOS, nitric oxide synthase; NPR-A, natriuretic peptide receptor A; NPR-B, natriuretic peptide receptor B; NPR-C, natriuretic peptide receptor C; PDE3, phosphodiesterase 3; PI3K, phosphoinositide-3-kinase; PKA, protein kinase A; PKC, protein kinase C; PKG, protein kinase G; PPARγ, peroxisome proliferator-activated receptor γ; sarcoKATP, sarcolemmal ATPdependent potassium channel. On the arrows «+» shows activation, and «-» blocking the pathways.
In 2000, corin was discovered, a transmembrane cardiac serine protease, which converts pro-ANP to ANP. 34 Corin was found in cardiomyocytes, atriums, ventricles of the human heart. 35 Proprotein convertase subtilisin/kexin-6 (PCSK6, also named PACE4) is enzyme converting pro-corin to corin, 36 and is expressed in the atrium and left ventricle of the heart. 37 Furin is another enzyme which converts pro-NPs into active NPs. 38 It is also expressed in the myocardium. 38 NPs are metabolized by the neutral endopeptidase neprilysin, which is expressed in many organs and tissues. 39 It is believed that selective neprilysin inhibitors may be effective in treatment of hypertension and heart failure (HF). 39
An increase in the secretion of NPs by cardiomyocytes occurs in response to an increase in the intracellular concentration of Ca2+. 40 The key role in this process is played by calmodulin kinase II. 40 In addition, it was found that infusions of norepinephrine evoked a 7-fold increase in the plasma ANP level in rabbits. 41 However, stimulation of right inferior cervical ganglion in vagotomized, anesthetized rabbits had no effect on the ANP level. 41 Norepinephrine and epinephrine also stimulate BNP release from the isolated human atrial myocardium. 42 Adenosine α1 and α2-adrenergic receptor (AR) agonists stimulate ANP secretion, whereas β1-AR stimulation inhibits ANP release. 43 BNP synthesis and secretion of by isolated cardiomyocytes is enhanced in response to hypoxia. 44,45 Chronic hypoxia causes an increase in ANP level in plasma of rats. 46 There are many stimuli that are known to increase gene expression and/or to trigger the release of CNP are pertinent to cardiovascular diseases, including shear stress, inflammatory cytokines such as tumor necrosis factor-α, interleukin 1β, transforming growth factor-β, and bacterial lipopolysaccharide. 26 It was found that the expression of ANP, NPR-A, and NPR-C is increased in endothelial NO-synthase deficient mice (eNOS-/-). 47 This may be a response to an increase in BP in these mice. However, an increase in NP synthesis can also be observed in the absence of an increase in BP, for example, in mice after bilateral renal ischemia-reperfusion. 48 A decrease in eNOS activity in the myocardium, cardiac hypertrophy, and an increase in BNP synthesis in the myocardium were observed 4 to 5 months after renal ischemia-reperfusion in mice. 48 To finally resolve the question of the role of NO in the regulation of NP synthesis, studies on isolated cardiomyocytes are necessary. The synthesis and secretion of BNP by isolated cardiomyocytes is enhanced in response to hypoxia. 44,45 Chronic hypoxia causes an increase in ANP level in plasma of rats. 46
Data From Experimental Studies
Infarct-Reducing Effect of Natriuretic Peptides
In a study performed on the isolated perfused rat heart, using coronary artery occlusion (35 min) and reperfusion (2 h), BNP (10-8 M) contributed to a striking reduction in infarct size by about 50% (Table 1). 49 This BNP effect was accompanied by a 2.5-fold increase in the cGMP level. Moreover, it was found that the inhibitor of mitochondrial ATP-sensitive K+ channel (mitoKATP channel) 5-hydroxydecanoate eliminates the cardioprotective effect of BNP. It is well known that cGMP and mitoKATP channel opening protect the heart from ischemic and reperfusion injuries. 50 Consequently, it can be assumed that the infarction-limiting effect of BNP is associated with mitoKATP channel opening in cardiomyocytes and an increase in the cGMP level in the myocardium. In a study performed in the isolated perfused rat hearts, using coronary artery occlusion (20 min) and reperfusion (120 min) to induce ischemia-reperfusion damage, synthetic human α-ANP (carperitide) reduced the infarct size/area at risk (IS/AAR) ratio by 40% (Table 1). 51 Later, in experiments on the isolated heart, the infarct-reducing effect of cANP4-23 and CNP was shown (Table 1). 18 In another study in the isolated perfused rat heart, BNP added to the perfusion solution 5 min before reperfusion caused a decrease in the IS/AAR ratio by 44% after local ischemia (35 min), followed by reperfusion (120 min) (Table 1). 52 In the latter study, it was found that the inhibition of NO synthase, blockade of sarcolemmal KATP channel (sarcKATP channel), and blockade of the mitoKATP channel led to the elimination of the infarction-reducing effect of BNP. The investigators concluded that the cardioprotective effect of BNP is associated with KATP channel opening and NO-synthase activation.
The Infarct-Reducing and Cytoprotective Effects of Natriuretic Peptides In Vitro.

Note. IS/AAR, infarct size/area at risk.
NPs exhibit cardioprotective properties not only in vitro, but also in vivo. Coronary artery occlusion (90 min) and reperfusion (6 h) were induced in dogs (Table 2). 53 Carperitide (human α-ANP) was administered (0.2 μg/kg/min) 15 min after coronary artery occlusion and infusion was continued until the end of reperfusion. Carperitide treatment contributed to a 84% reduction in infarct size. It was found that intravenous bolus administration of rabbit ANP (2.5 μg/kg) 15 min before ischemia induced a 84% reduction in the IS/AAR ratio in rabbits with coronary occlusion (90 min) and reperfusion (3 h) (Table 2). 54 In a dog model, myocardial ischemia was simulated by reducing blood flow in the left anterior descending coronary artery by 66% (Table 2). 27 Carperitide was infused (0.1 μg/kg/min) intracoronary during cardiac ischemia. It turned out that carperitide improved contractility of the heart and increased coronary blood flow by enhancing cGMP synthesis. HS-142 -1, a NPR antagonist, eliminated the protective effects of carperitide. In addition, the NO-synthase (NOS) inhibitor L-NAME eliminated carperitide’s protective effects. Carperitide reduced the IS/AAR ratio, which was determined after ischemia (90 min) and reperfusion. These data indicate that carperitide increases coronary blood flow and cardiac tolerance to ischemia/reperfusion. 27 In another study, BNP was administered to rats with permanent coronary occlusion for 28 days (Table 3). 55 Half of the rats in the untreated infarction control group died during this period. At the end of the experiment, the left ventricular (LV) weight increased by 28%, and left ventricular end-diastolic pressure (LV EDP) increased 5-fold in untreated infarction control rats. Chronic BNP treatment reduced EDP by 46%, decreased LV weight by 17%, and increased the survival of rats with myocardial infarction. The authors concluded that chronic administration of BNP increases survival of rats with permanent coronary occlusion and prevents postinfarction remodeling of the heart. 27 A study performed in ANP-null mice with coronary artery occlusion (30 min) and reperfusion (120 min) showed that lack of endogenous ANP aggravates adverse cardiac remodeling induced by pressure overload in mice. 56 Mice lacking NPR-A gene Npr1 (Npr-/-) have marked cardiac hypertrophy and fibrosis disproportionate to their increased blood pressure. 57 Recently, a study has been published in cardiomyocyte (cmCNP-/-), endothelium (ecCNP-/-), and fibroblast (fbCNP-/-)-specific CNP knockout mice, as well as global (NPR)-B-/-, and NPR-C-/- knockout mice. 33 Global ischemia of the isolated murine heart evoked an increase in infarct size and diminished functional recovery in cmCNP-/- and NPR-C-/-, but not in ecCNP-/- mice vs. wild-type controls. The cardiac phenotype of cmCNP-/-, fbCNP-/-, and NPR-C-/- (but not ecCNP-/- or NPR-B-/-) mice was more sensitive to pressure overload- and sympathetic hyperactivation-induced HF compared with wild-type mice. 33
The Infarct-Reducing Effect of Natriuretic Peptides In Vivo.

Note. IS/AAR, infarct size/area at risk.
Natriuretic Peptides Prevent Post-Infarction Cardiac Remodeling.
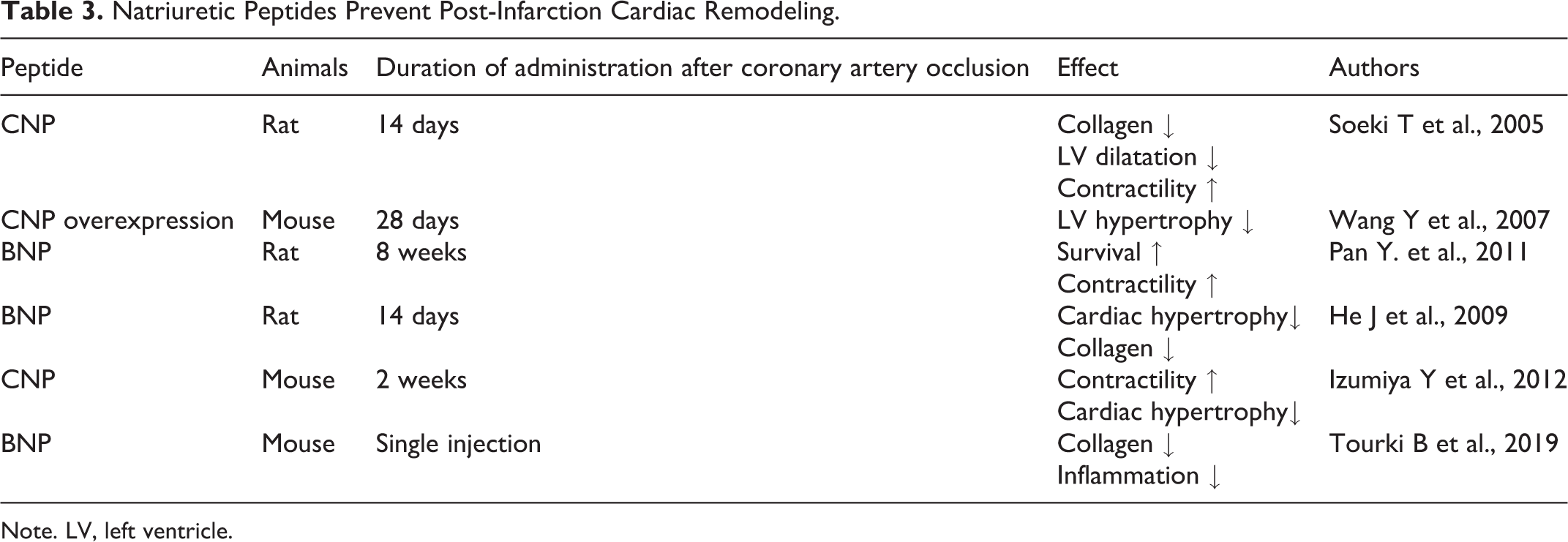
Note. LV, left ventricle.
The use of neprilysin inhibitors may be another approach to prevent post-infarction cardiac remodeling. It was shown that the neprilysin inhibitor LCZ696 can increase the survival of mice with experimental myocardial infarction treated for 28 days. 58 Consequently, NPs inhibit adverse cardiac remodeling in different species and experimental settings. However, it remains unclear which NPRs are involved in the antifibrotic effects of NPs. It is well known that transforming growth factor-β1 (TGF-ß1) plays an important role in cardiac fibrosis. In isolated fibroblasts, knockdown of NPR-C resulted in a marked reduction of collagen in murine atrial and ventricular fibroblasts/myofibroblasts in response to TGF-ß1 stimulation. 59 There was a marked reduction in atrial fibrosis gene expression in mice with NPR-C knockdown and transverse aortic constriction (TAC) compared to wild-type mice with TAC. These data indicate that NPR-C may play a beneficial role in prevention of cardiac remodeling.
One of the most widespread diseases of our time is diabetes mellitus (DM), which exacerbates the course of cardiovascular diseases. Therefore, it was important to find out how it affects the cardioprotective effect of ANP. The study was performed in rats with alloxan-induced diabetes, 60 and showed that ANP did not have an infarct-limiting effect in rats with diabetes (Table 2). It should be noted that alloxan treatment mimics type 1 diabetes, while type 2 diabetes prevails among patients with diabetes. Therefore, the latter study does not rule out that ANP may have cardioprotective effects in patients with type 2 diabetes. It has been suggested that BNP may be involved in the pathogenesis of diabetic cardiomyopathy. 61 There are data that diabetes promotes an increase in ANP and BNP expression in the heart of streptozotocin-induced diabetic rats. 62 However, it is unclear whether enhancement of ANP and BNP expression is protective or aggravates diabetic cardiomyopathy. No differences were found between patients with acute heart failure with diabetes and without in terms of the serum BNP level. 63 It is established that the sodium-glucose cotransporter 2 (SGLT2) inhibitors reduce cardiovascular mortality and re-hospitalization for heart failure (HF) in patients with type 2 diabetes (T2D). 64,65 In addition, the SGLT2 inhibitor empagliflozin prevented post-infarction cardiac remodeling in rats. 66 However, in a randomized, placebo-controlled, double-blind, multicentre pilot study including 80 acute HF patients with and without diabetes a positive effect of empagliflozin on HF and on plasma Pro-BNP levels could not be detected. 67 In contrast, in a multicentre, parallel-group phase IIA study including 125 patients with T2DM and HF it was found that the combined SGLT1/SGLT2 inhibitor licogliflozin reduced the plasma NT-proBNP level vs placebo, whereas empagliflozin had no effect on the NT-proBNP level. 68 The EMMY trial is designed to finally clarify the appropriateness of empagliflozin for the treatment of HF. 69
It has been well established that NPs can prevent cardiac reperfusion injury even when they are used after the ischemic injury of the heart has already occurred. 52,70 -74 Isolated rabbit hearts were subjected to a 30-min coronary artery occlusion, followed by a 120-min reperfusion. 70 When ANP was added to the perfusion solution 5 min before reperfusion, it caused a decrease in the IS/AAR ratio by 60%. Similarly, in studies performed in isolated perfused rat hearts, BNP and ANP prevented cardiac reperfusion injury after coronary occlusion and improved left ventricular function after cardiac global ischemia-reperfusion when added to the perfusion solution (Table 1). 52,71,73,75 In concordance with the ex vivo studies, in vivo studies in rats subjected to coronary occlusion (30 min) and reperfusion (4 h) showed that infusion of BNP (0.03 μg/kg min, i.v.), starting 15 min before reperfusion until the end of reperfusion, decreased the IS/AAR ratio by approximately 50%. 72 In line with the latter study, it was shown that intraperitoneal administration of BNP 5 min before reperfusion contributed to a decrease in the IS/AAR ratio by an average of 50%. 71
Natriuretic Peptides and Postinfarction Myocardial Remodeling
There is good evidence that NP treatment has beneficial effects on postinfarction cardiac remodeling. For example, it was found that continuous CNP infusion (0.1 µg/kg/min) by osmotic mini-pump during 2 weeks after permanent coronary artery occlusion prevented post-infarction myocardial remodeling (Table 3). 76 Furthermore, Wang and coworkers showed that cardiomyocyte-specific CNP overexpression had no effect on infarct size in mice, but prevented both left and right ventricular hypertrophy after coronary artery occlusion (Table 3). 77 When rats with permanent coronary occlusion were injected intravenously with BNP (15 μg/kg/day) over 8 weeks, BNP treatment prevented cardiac hypertrophy and a decline in ejection fraction, decreased the plasma angiotensin II level, and decreased collagen content in the myocardium (Table 3). 78 In an experiment in which angiotensin II was continuously infused subcutaneously into mice by osmotic minipump during 2 weeks, co-infusion of CNP prevented angiotensin II-induced myocardial hypertrophy, cardiomyocyte hypertrophy, interstitial fibrosis (Table 3). 79 In addition, CNP decreased angiotesin II-induced cardiac superoxide generation by myocardial tissue. 79 The latter authors concluded that CNP suppresses angiotensin-induced cardiac hypertrophy by inhibition of superoxide production.
It is well known that one of the signs of post-infarction remodeling in the heart is myocardial fibrosis. In this context it was found that a single injection of BNP before the restoration of coronary blood flow reduced the myocardial collagen level by about 30%, 14 days after reperfusion (Table 3). 74 It is well known that inflammation plays an important role in ischemia/reperfusion injury of the myocardium. 80,81 Therefore, it has been suggested that the cardioprotective effect of BNP during reperfusion may be a consequence of the limitation of leukocyte infiltration in the infarcted area. Indeed, it turned out that after 2 days of cardiac reperfusion, the number of leukocytes in the infarction zone in BNP-treated rats decreased by 30% (Table 2). 74 These data indicate that BNP prevents cardiac reperfusion injury, and reduces the manifestations of postischemic fibrosis of the heart, possibly through the limitation of leukocyte infiltration into the necrosis zone. The cardioprotective effect of BNP is long-term in nature and persists for at least 14 days.
After genetic knockout of the Npr1 gene encoding NPR-A in mice, blood pressure rises in mice and cardiac hypertrophy develops. 24 BP was elevated by 41 mm Hg in Npr1-/- mice, together with 60% increased heart weight/body weight ratio and cardiomyocyte hypertrophy. 30 These data indicate that endogenous NPs can prevent the development of cardiac hypertrophy. Furthermore, it was demonstrated that cardiac-specific disturbance of NPR-A activity accentuates adverse cardiac remodeling and mortality in response to pressure overload in mice. 82 In contrast, subcutaneous infusion of CD-NP (synthetic NP) during 2 weeks prevents left ventricular fibrosis and decreased circulating aldosterone levels in rats with unilateral nephrectomy. 83 Another study of the same group examined the role of corin and furin in atrial fibrosis in dogs with rapid right ventricular pacing for 10 days. 84 In the left atria, corin mRNA and protein expression were lower, whereas furin mRNA and protein expression were higher than normal. 38 Moreover, it was demonstrated that the failing human left ventricle is characterized by increased fibrosis and reduced CNP gene expression. 84
Natriuretic Peptides, Apoptosis, Pyroptosis and Autophagy
In 2009, Wu et al demonstrated that BNP can protect the heart from ischemia-reperfusion injury by inhibition of apoptosis. 85 The latter study was performed in rabbits subjected to coronary occlusion (45 min) and reperfusion (3 h). BNP infusion was started 5 min before reperfusion and continued throughout reperfusion. BNP infusion reduced infarct size by 44% (Table 2), and the number of apoptotic cells (TUNEL-positive cardiomyocytes) in the heart by approximately 60%. 85 At the same time, BNP increased protein expression of anti-apoptotic Bcl-2, reduced pro-apoptotic Bax protein expression, and prevented caspase-3 activation. Similar data were obtained by other researchers. 86 BNP was also shown to prevent hypoxia-induced apoptosis of neonatal rat cardiomyocytes. 87 BNP prevented the fall of Δψ in mitochondria, and decreased ROS generation in cardiomyocytes. In addition, BNP increased Bcl-2, decreased the Bax level, and decreased the Smac/DIABLO ratio. 87 The PI3 kinase inhibitor wortmannin abolished the anti-apoptotic effect of BNP. 58 Furthermore, is was reported that BNP prevented apoptosis of H9c2 cardiomyoblasts under hypoxia/reoxygenation conditions. 88 Pretreatment with the selective cGMP-dependent protein kinase G (PKG) inhibitor KT-5823 significantly weakened the anti-apoptotic effect of BNP. 88 In contrast, some experiments suggested that BNP can actually enhance the apoptosis of neonatal rat cardiomyocytes in conditions of moderate hypoxia and increase caspase-3 activity. 89
It is well known that caspase-1, interleukin-1β (IL-1β) and inflammasome formation are involved in the development of pyroptosis. 90 In this context, it has been shown that ANP and BNP are able to suppress lipopolysaccharide-induced IL-1β release from human monocytes, and to inhibit inflammasome formation and caspase-1 activation in monocytes. 91,92 Consequently, ANP and BNP can suppress pyroptosis of monocytes. Whether NPs might inhibit pyroptosis of cardiomyocytes is currently unknown.
It is well known that mTOR (mammalian target of rapamycin) kinase is a strong inhibitor of autophagy. 93 Therefore, using the mTOR inhibitor rapamycin, it is possible to indirectly estimate a role of autophagy in a particular process. It was found that BNP inhibits mTORC1 in adipocytes. 94 Moreover, pretreatment with rapamycin abolished the infarct-limiting effect of BNP-mediated postconditioning. 72 These data indirectly indicate that the infarct-reducing effect of natriuretic peptides may depend on modulation of autophagy. However, these indirect findings require confirmation.
Natriuretic Peptides and ROS
It is well known that a high content of reactive oxygen species (ROS) induces cardiomyocyte injury, whereas in small concentrations ROS can function as intracellular signaling molecules. 95,96 Therefore, it was important to find out how natriuretic peptides affect the ROS level in cardiomyocytes. Angiotensin II was infused subcutaneously into mice using an osmotic minipump over 2 weeks to stimulate ROS production in the heart in vivo. 79 Co-administration of CNP decreased angiotensin II-induced cardiac hypertrophy and superoxide production in myocardial tissue. This effect was accompanied by suppression of NADPH-oxidase expression. 79 These data indicate that CNP attenuated angiotensin II-induced cardiac hypertrophy and fibrosis, the effect which was associated with reduced superoxide radical production. Similarly, treatment of adult spontaneously hypertensive rats (SHRs) with an ANP derivative for 2 weeks attenuated hypertension, and restored the impaired vasorelaxation toward control level. 25 In addition, ANP reduced the superoxide anion and peroxynitrite levels, and decreased NADPH-oxidase activity in aorta, heart, and kidney of SHRs. The latter authors used C-atrial natriuretic peptide (cANP)4-23, a peptide with selectivity to NPR-C. This fact allowed them to conclude that selective stimulation of NPR-C without an increase in the cGMP level can prevent the development of hypertension and an increase in ROS production. 25 Along similar lines, it was shown that ANP reduced lipopolysaccharide-induced intracellular ROS production in endothelial cells, 97 that BNP inhibits angiotensin II-induced ROS production in pulmonary arterial smooth muscle cells 67 and postnatal ductus arteriosus smooth muscle cells, 98 and that ANP prevents isoproterenol-induced mitochondrial ROS production in failing cardiomyocytes. 99 In 2018, Jain A et al demonstrated that cANP4-23 inhibits superoxide generation and NADPH oxidase activity in vascular smooth muscle cells (VSMC) isolated from SHRs and Wistar rats. 100 The ability of cANP4-23 to inhibit the enhanced oxidative stress in VSMC isolated from SHRs was confirmed in other studies. 101,102
High frequency stimulation increased ANP secretion in atria of both diabetic and sham rats. Interestingly, the ROS scavenger N-acetyl-cysteine (NAC) attenuated but did not abolish ANP secretion, whereas the NADPH inhibitor apocynin had no effect in sham rat atria. However, both apocynin and NAC completely inhibited ANP secretion in diabetic rat atria. 103 These data indicate that ROS can trigger ANP secretion especially in diabetic rats.
Natriuretic peptides can not only inhibit ROS production but also stimulate ROS generation in some experimental settings. In this context, it was demonstrated that CNP enhances NADPH-depended ROS production in rat aorta homogenates. 104 It is well known that ROS play a role as intracellular messengers in cardioprotective pre- and post-conditioning effects. 105 For example, the infarct-reducing effect of bradykinin is mediated via ROS generation in the myocardium. 106 Although the majority of studies suggest the inhibitory effect of NPs on ROS production in the strained heart, the exact role of the modulation of ROS production in the infarct-limiting effect of NPs is currently not known.
In conclusion, there is solid evidence from experimental studies that natriuretic peptides limit infarct size, increase survival of animals after experimental myocardial infarction, prevent cardiac reperfusion injury, have anti-inflammatory effects, and prevent post-infarction remodeling of the heart. In addition, it was established that BNP prevents hypoxia-induced apoptosis in cardiomyocytes. Indirect evidence suggests that natriuretic peptides may inhibit pyroptosis and may stimulate autophagy. Currently, there is only little information on how natriuretic peptides affect necroptosis, autophagy and pyroptosis of cardiomyocytes under conditions of hypoxia/reoxygenation. Taken together, the findings from experimental studies strongly suggest that natriuretic peptides or NPR receptor agonists may be useful in the treatment of myocardial infarction. Thus, most studies show that natriuretic peptides inhibit ROS production in cardiomyocytes, aorta, heart, kidney and VSMC.
Data From Clinical Studies
Two multicenter, prospective, randomized, blind, placebo-controlled trials included AMI patients who were given intravenous infusion of placebo or ANP (0.025 µg/kg/min) 3 days after hospitalization (Table 4). 107 Infarct size was estimated indirectly by the creatine kinase (CK) level in the blood. It turned out that the CK level was lower in patients who were injected with ANP. ANP treatment reduced infarct size by 14.7%. Six to 12 months after AMI, the LV ejection fraction (LV EF) was greater in patients who were injected with ANP. In 2015, the results of a study in patients with ST-segment elevation myocardial infarction (STEMI) and PCI were published (Table 4). 108 BNP was first administered intravenously by a bolus (1.5 μg/kg) before PCI, and then 0.0075 μg/ kg/min was infused intravenously for 120 hours. The infarction was estimated indirectly by the level of the serum markers of cardiomyocyte necrosis, troponin I and CK-MB. Echocardiography was performed 7 days later and 6 months after myocardial infarction. The serum troponin I and CK-MB levels were lower in patients who were administered BNP compared with the control group (nitroglycerin infusion). Although there were no differences between these groups in LV EF after 7 days, LV EF was higher in patients who were infused with BNP, 6 months after myocardial infarction. The authors concluded that intravenous administration of BNP contributes to limit the size of myocardial infarction and to inhibit postinfarction remodeling of the heart. 108
Natriuretic Peptides in Treatment of Myocardial Infarction.
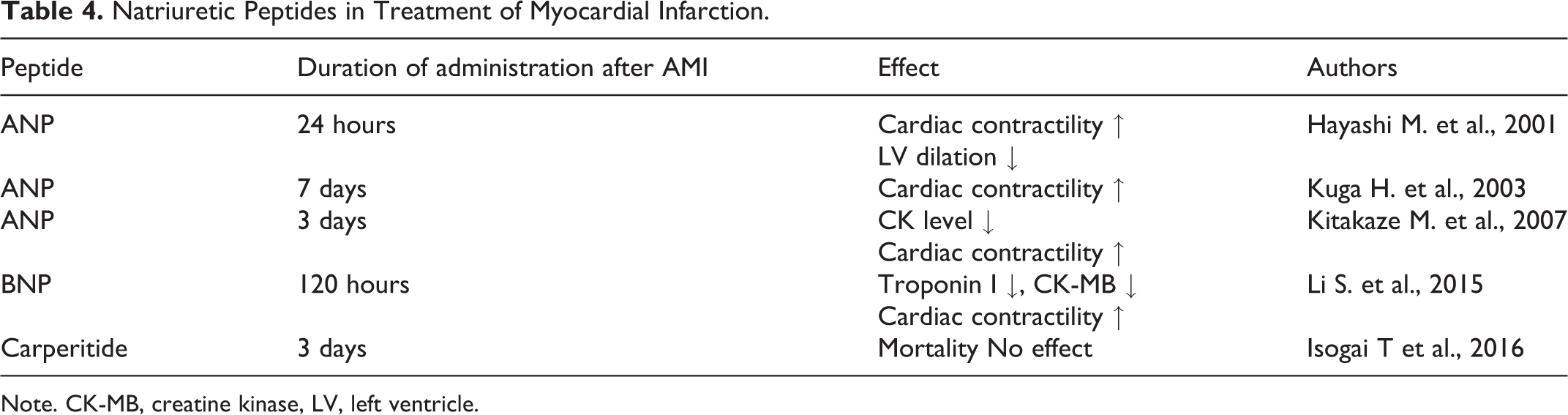
Note. CK-MB, creatine kinase, LV, left ventricle.
In a placebo-controlled study including 133 patients with coronary artery bypass grafting and cardioplegic cardiac arrest, which is essentially cardiac ischemia, human ANP was infused with 0.02 μg/kg/min immediately after the initiation of cardiopulmonary bypass. 109 The incidence of postoperative complications was lower in patients who were infused with ANP. In addition, LVEF in the postoperative period was higher after administration of ANP, and circulating BNP levels as an indicator of heart failure were lower. 109 However, there was no difference between the groups in mortality rate in the first year post-operatively.
There is also evidence that ANP has beneficial effects on postinfarction remodeling of the heart. ANP was infused continuously (0.025 μg/kg/min) for 24 hours in patients with STEMI and PCI (Table 4). 110 The control group included patients with AMI who were injected nitroglycerin. One month after AMI, LVEF was higher in patients who were injected with ANP. In addition, patients treated with ANP had less LV dilatation, and lower plasma levels of aldosterone, angiotensin II, and endothelin-1. The authors concluded that ANP inhibits postinfarction remodeling of the heart. 110 In another study, human ANP was infused continuously (0.025 μg/kg/min) for 7 days in patients with STEMI and PCI (Table 4). 111 In addition, ANP was administered intracoronary during PCI. Six months after myocardial infarction, the LV end-diastolic volume (LV EDV) was lower, and LV EF was higher in patients who were injected with ANP. These findings suggest that ANP may prevent postinfarction remodeling of the heart.
The most important indicator of the efficacy of drug therapy in any disease is the reduction in mortality. In 2016, the results of a multicenter study performed in Japan were published. 112 The study included 60,592 patients with AMI and PCI (81,89 ANP group, 52,403 control group) from 850 hospitals. Investigators were not able to detect a significant difference in 30-day mortality between the ANP (carperitide) and control group (3.4% vs. 3.8%, respectively). Carperitide was administered on the second day after PCI. Perhaps for this reason, the positive effect of carperitide could not be detected. In addition, this study was not the randomized, blind, and placebo-controlled trial. 112 Hence, it is unclear whether a study by Isogai and coworkers can provide conclusive evidence of a lacking effect of ANP on mortality after AMI (Table 4).
Thus, natriuretic peptides increase tolerance of the human heart to ischemia/reperfusion and have the infarct-limited effect in patients with AMI. ANP prevents post-infarction remodeling of the heart.
Natriuretic Peptides as Predictors of Mortality
It was reported that an increase in BNP levels in blood of cardiosurgical patients before cardiopulmonary bypass, which is accompanied by total cardiac ischemia, may predict death of patients. 113 Similar data were obtained regarding an increase in the N-terminal pro BNP (NT-ProBNP) level in the period preceding cardiac surgery. 114 It was shown that the elevated NT-proBNP level during admission of patients with AMI was associated with a higher risk of mortality. 115 An increase in the N-terminal pro-BNP level predicts in-hospital mortality in patients with heart failure. 116 Based on these data, it may be concluded that endogenous NPs actually exacerbate ischemia/reperfusion cardiac injury. However, it has to be considered that increased circulating levels of BNP and NT-proBNP are markers of pre-existent cardiac hypertrophy and heart failure. ANP and BNP are powerful clinical markers of cardiac hypertrophy and cardiac dysfunction. 40 Hence, the abovementioned associations between elevated circulating levels of endogenous BNP and negative outcome do not allow any firm conclusions about the potential therapeutic efficacy of NPs to limit ischemia/reperfusion injury in the heart. In our opinion, increasing levels of BNP and NT-proBNP is a protective response to extreme effects such as AMI and surgery. Indeed, there is evidence that the plasma ANP level increases in humans by 3-fold during stress exposure. 117 We believe that NPs protect the heart from adverse effects.
Potential Mechanisms Underlying the Infarct-Reducing and Anti-Fibrotic Effects of Natriuretic Peptides
Anti-Inflammatory Effect of Natriuretic Peptides
It was found that inflammation plays an important role in reperfusion injury to the heart and the development of myocardial fibrosis. 118,119 Therefore, it was important to evaluate the effect of natriuretic peptides on inflammation during ischemia/reperfusion of the heart. It was found that ANP limits polymorphonuclear neutrophils adhesion to hypoxic endothelial cells. 120 It has been found that ANP may protect the heart from angiotensin II-induced myocardial remodeling by attenuating inflammation. 121 Pretreatment with ANP reduced TNF-α-induced expression of monocyte chemoattractant protein-1 (MCP-1) by human umbilical vein endothelial cells. 122 It has been established that ANP prevents an increase in the serum tumor necrosis factor-α (TNF-α) level in mice after bacterial lipopolysaccharide administration but had no effect on TNF-α expression in the mouse heart. 123 However, there are data that BNP had no effect on TNF-α release from macrophages. 124
It was demonstrated that plasma concentrations of proinflammatory cytokines TNF-α, interleukin-6 (IL-6), and interleukin-1 (IL-1) is elevated in Npr1-/- mice in comparison with wild-type mice. 125 It was shown that ameliorates lipopolysaccharide-induced acute lung injury and reduced the serum IL-1β and IL-6 levels in rats. 126 Investigators showed that oleic acid-induced acute lung injury is associated with an increase in the TNF-α, IL-1β and IL-6 levels in rats. 127 Pretreatment with ANP prevented this elevation of the proinflammatory cytokines level. Other investigators showed that ANP prevents elevation of the serum TNF-α and IL-6 levels in dogs after lipopolysaccharide administration. 128 High mobility group box 1 protein (HMGB1) released by necrotic cells and stimulates proinflammatory cytokine production positively activated macrophages and monocytes. 129 BNP was administrated 15 min before cardiac reperfusion. 71 Authors demonstrated that BNP decreases infarct size, reduces TNF-α, IL-6, HMGB1 content in myocardial tissue. It was established that intravenous administration of CNP (25 μg/kg) to rats with hemorrhagic shock prevents an increase in TNF-α, IL-1β and IL-6 content in the kidney. 130 Investigators found that CNP reduces secretion of MCP-1 and plasminogen activator inhibitor-1 (PAI-1) by cardiac fibroblasts. 131 They suggest that a decrease in MCP-1 and PAI-1 secretion is involved in the antifibrotic effect of the peptide. It was demonstrated that CNP prevents ventricular hypertrophy in SHR and induces an anti-inflammatory and antifibrotic response in SHR. 132 Other investigators also report on the anti-inflammatory effect of CNP. 26
Thus, a number of studies have shown that natriuretic peptides have the anti-inflammatory effect, reduce the inflammatory mediators’ level in the blood and tissues. However, it remains unclear to what extent the anti-inflammatory effect of these peptides is involved in their infarct-limiting and the antifibrotic effects.
Natriuretic Peptides, Angiotensin II, Aldosterone, Endothelin-1
It is well known that angiotensin II, endothelin-I, aldosterone are involved in the postinfarction adverse remodeling heart. 118,133 The AT1 receptor antagonists contribute to a decrease in the IS/AAR ratio. 134,135 The endothelin receptor antagonists have the cardioprotective effect on coronary artery occlusion and reperfusion. 136 Therefore, it was important to evaluate how these peptides affect the angiotensin II and aldosterone levels. It was shown that ANP inhibits ACTH-stimulated aldosterone secretion in mice. 137 ANP infusion promoted a decrease in the plasma aldosterone and angiotensin II levels in patients with AMI. 110 Angiotensin II-induced ventricular hypertrophy is suppressed in mice with BNP overexpression. 138 The plasma angiotensin II level increased 4-fold 8 weeks after the experimental myocardial infarction in rats. The course BNP administration led to a decrease in the angiotensin II level almost to normal value. 78 It was shown that the angiotensin II induces cardiac fibroblast proliferation and collagen synthesis by these cells. 139 ANP inhibited both effects of angiotensin II. siRNA inhibited NPR-A expression in isolated cardiac fibroblasts and eliminated the effects of ANP. 139 Consequently, NPR-A abolishes the effects of angiotensin II on the cell. In response to 21 days of transverse aortic constriction, NPR-1-/- mice developed enhanced left ventricular hypertrophy, fibrosis and contractile dysfunction. 140 Pretreatment with the mineralocorticoid receptor (MR) selective antagonist eplerenone prevented cardiac hypertrophy. Authors demonstrated coimmunoprecipitation MRs and ANPs. Authors proposed that aldosterone can cause a conformational change of MR/NPR-A complex which was prevented by ANP. 140 We have already reported that long-term CNP infusion attenuates angiotensin II-induced cardiac superoxide production and myocardial remodeling. 79 It has been demonstrated that ANP may protect the heart from angiotensin II-induced myocardial remodeling by attenuating inflammation. 121 It was demonstrated that ANP attenuated angiotensin II-induced aldosterone synthesis in a human adrenocortical cell line (NCI-H295 R). 141
In 1993, Uemasu J et al demonstrated that ANP infusion induced a decrease in the plasma endothelin-1 level in men. 142 Another group investigators found that ANP suppresses endothelin-1 gene expression and proliferation in cardiac fibroblasts. 143 Administration of ANP suppressed the plasma endothelin level in patients with AMI. 110 Investigators established that in vitro, angiotensin II increased endothelin-1 expression in cardiac fibroblasts, which were reduced by ANP. 121
Presented data showed that natriuretic peptides can suppress aldosterone, angiotensin II, endothelin-1 synthesize and secretion and inhibit the effects aldosterone, angiotensin II on the post-receptor level through intracellular signaling events.
Natriuretic Peptides, cAMP, cGMP, PKG
The cardiovascular effects of atrial natriuretic peptides are mediated through intracellular signaling mechanisms, the most important of which are cAMP and cGMP. It was demonstrated that cGMP and PKG provide cardiac tolerance to ischemia/reperfusion. 144 In 2006, Yang et al., found that the soluble guanylyl cyclase inhibitor ODQ eliminates the infarct limiting effect of ANP at cardiac reperfusion. 70 In 2010, Qvigstad et al found that CNP enhances β1-adrenergic receptor mediated increase of cAMP levels in failing hearts through phosphodiesterase 3 (PDE-3) inhibition. 145 Investigators demonstrated that BNP increases the cAMP level in PC12 cells and induces protein kinase A (PKA) phosphorylation in these cells. 146,147 They showed that BNP inhibited phosphodiesterase type 3-mediated cAMP hydrolysis in PC12 cells. It can be assumed that NPs can increase in cAMP content in cells via inhibition of PDE3 that could be detrimental in heart failure due to an increase in adrenergic drive. Protein kinase G is involved in this effect of CNP. In 2014, Vellaichamy E et al demonstrated that in Npr1(-/-) mice the myocardial guanylyl cyclase activity and the cGMP level in the heart were reduced by 96% and 87%, respectively. 30 However, these indices were elevated by 2.8-fold and 3.8-fold, respectively, in Npr1(++/++) mice. 30 CNP increased cGMP and enhanced β1- and β2-AR-mediated inotropic and lusitropic responses, in non-failing and failing hearts. 148 BNP increased cGMP, but did not affect inotropic or lusitropic responses on the AR agonists, indicating different compartmentation of cGMP from different NPRs. 148 It has been shown that a low concentration of norepinephrine increases cAMP content in isolated left ventricular cardiomyocytes. 148 Consecutive addition of CNP induced a further increase in the cAMP level in cardiomyocytes. The authors suggest that this CNP effect is associated with increased cGMP-mediated phosphodiesterase (PDE) 3 inhibition by NPR-B receptor activation. 148
Thus, the presented data indicate that natriuretic peptides activate guanylyl cyclase, activate PKG and PKA, and reduce the PDE-3 activity (Figure 3). It is unclear which of these changes provide increased cardiac tolerance to ischemia/reperfusion. It is well known that the β1-adrenergic receptor (β1-AR) antagonists inhibit cAMP synthesis and enhance cardiac tolerance to ischemia/ reperfusion. 149 However, there is evidence that β1-AR stimulation enhances tolerance of this organ to ischemia/reperfusion. 150
Natriuretic Peptides and Protein Kinases
It is well known that kinases play an important role in intracellular signaling and regulation of cardiac tolerance to ischemia/reperfusion. 105,151 So, it was established that the activation of a number of kinases (AMPK, PKC, PI3 K, Akt, ERK1/2, PKG) increases the resistance of the heart to ischemia and reperfusion. 105,151 At the same time, increased activity of glycogen synthase kinase 3β (GSK-3β) contributes to the occurrence of ischemic and reperfusion injury to the heart. 105,151 Inhibition of protein kinase C (PKC) contributed to the disappearance of the cardioprotective effect of carperitide. 51 The hepatoprotective effect of cANP4-23 during hypoxia of liver is mediated via PKC-δ activation. 152 There are evidence that ANP-induced endothelial cells proliferation in vitro is mediated via activation of PKG, PI3 K, Akt, ERK1/2. 153 In 2006, Yang et al. found that the soluble guanylyl cyclase inhibitor ODQ eliminates the infarct limiting effect of ANP at cardiac reperfusion. 70 It was demonstrated that wortmannin, a phosphatidylinositol-3-kinase (PI3 K) inhibitor, or PD98059, a MEK kinase and extracellular signal-regulated protein kinases (ERK1/2) inhibitor, also abolished the infarct-reducing effect of ANP at reperfusion of the heart. 70 Consequently, the cardioprotective effect of ANP is mediated via the activation of PI3 K, MEK/ERK (ERK1/2). It was found that cANP4-23 inhibits VSMC proliferation via MEK/PI3K/Akt pathway. 154 There are data that ANP prevents mitochondrial permeability transition (MPT) pore opening in cardiac H9c2 cells via inhibition GSK-3β and activation of PKG and PI3 K. 155 It was shown that the PI3 K inhibitor LY294002 completely abolished the cardioprotective effect of BNP at reperfusion of the heart. 71 Investigators believe that PI3K/Akt signaling pathway is involved in anti-inflammatory effect of BNP. 71 In addition, investigators demonstrated that BNP increases the phospho-Akt (activated) kinase level in the myocardium. These facts indicated that PI3 K and Akt are involved in the infarct-limiting effect of BNP during reperfusion. CNP induced Akt phosphorylation in endothelial cells. 97 Pretreatment with inhibitors of PI3 K, Akt, p70s6 k kinases (wortmannin, SH-6, rapamycin) completely eliminated the infarct-reducing effect of BNP at reperfusion of the heart. 72 Authors concluded that the PI3K/Akt/p70s6 k pathway is involved in the protective effect BNP at reperfusion. In 2018, Zhang et al. demonstrated that BNP attenuates hypoxia-induced injury in H9c2 cardiomyocytes via stimulation of PI3K/Akt/mTOR pathway. 156 Investigators could established that long noncoding RNA EGOT is involved in activation of PI3K/Akt/mTOR pathway. It was found that CNP and cANP4-23 stimulates phosphorylation of AMP activated kinase a1 (AMPK) in tissue of isolated beating rat atria. 157 This effect of ANP and cANP4-23 was mediated via activation of PKG and Gi/o-proteins.
Consequently, the cardioprotective effect of natriuretic peptides is mediated via activation of AMPK, PKC, PI3 K, MEK/ERK (ERK1/2), p70s6 k, Akt and inhibition of GSK-3β (Figure 3).
Natriuretic Peptides and NO-Synthase
It is well known that NO-synthase is an important regulator of cardiac resistance to ischemia/reperfusion. 143 Investigators established that the infarct-reducing effect of BNP during reperfusion is mediated via NO-synthase. 52 The NOS inhibitor L-NAME abolished the infarct-reducing effect of carperitide 27 and ANP. 51,60 In 2010, Caniffi C et al. demonstrated that cANP4-23, a selective NPR-C receptor agonist, increased NOS activity in the heart. 158 cANP4-23 had no effect on NOS isoforms expression. The inhibitors of inducible NOS (iNOS) and neuronal NOS (nNOS) did not affect CNP-induced NOS activity. Cardiovascular NOS response to ANP4-23 was reduced by the Gi-protein inhibitor pertussis toxin and the calmodulin inhibitor calmidazolium. 158 Authors concluded that cANP4-23 interacts with NPR-C and activate Ca-calmodulin dependent eNOS via Gi protein.
Consequently, NOS is involved in the cardioprotective effect of natriuretic peptides (Figure 3). However, it remains unclear how the signal is transmitted from natriuretic peptide receptors on sarcolemma to NOS and soluble guanylyl cyclase in cytoplasm of the cell.
Natriuretic Peptides, HO-1 and PPARγ
It is known that hemeoxygenase-1 (HO-1) promotes an increase in cardiac tolerance to ischemia/reperfusion. 151 It has been established the stimulation of peroxisome proliferator-activated receptor γ (PPARγ) increases myocardial resistance to ischemia/reperfusion. 159 It was shown that CNP induced activation of HO-1 expression in HUVECs. 66 There is no data on the ability of natriuretic peptides to stimulate HO-1 expression in cardiomyocytes. It was shown that ANP activates PPARγ expression in the area at risk of the swine heart at ischemia/reperfusion. 160
The presented data indicate that HO-1 and PPARγ may be involved in the infarct-reducing effect of natriuretic peptides (Figure 3).
Natriuretic Peptides, KATP Channels and MPT Pore
It has been demonstrated that ATP-sensitive (KATP) channels opening promotes an increase in cardiac tolerance to ischemia/reperfusion. 105 Therefore, it was important to evaluate the role of these channels in the cardioprotective effect of natriuretic peptides. The blockade of mitochondrial KATP channel (mitoKATP) channel led to the disappearance of the cardioprotective effect of carperitide. 51 An involvement of mitoKATP in the cardioprotective effect of BNP has been demonstrated in D’Souza’s study. 49 It has been shown that the mitoKATP inhibitor 5-hydroxydecanoate eliminated the infarct-sparing effect of ANP at cardiac reperfusion. 70 Investigators established that the infarct-reducing effect of BNP during reperfusion is mediated via mitoKATP and sarcolemmal KATP (sarcKATP) channel opening. 52 In 2014, Baxter’s group demonstrated that cardioprotective effect of ANP, BNP and cANP4-23 is mediated via sarcKATP channel opening. 161
It has been established that MPT pore plays an important role in the cardioprotective effect of pre- and postconditioning. 105 MPT pore opining induces apoptosis of cardiomyocytes, MPT pore closing prevents ischemic/reperfusion injury of the heart. 162 It has been shown that the cardioprotective effect of BNP at cardiac reperfusion is mediated via MPT pore closing. 73 There are data that ANP prevents MPT pore opening in H9c2 cardiomyoblasts. 155 The presented data are indirect evidence that natriuretic peptides exhibit the anti-apoptotic effect.
Consequently, the cardioprotective effect of natriuretic peptides is mediated via sarcKATP channel and mitoKATP channel opening. The cardioprotective effect of BNP is mediated via MPT pore closing (Figure 3).
Natriuretic Peptides and TGF-β1
It was found that transforming growth factor-β1 (TGF-β1) plays an important role in the development of adverse postinfarction myocardial remodeling. 163 It has been established that long-term treatment with BNP prevents postinfarction myocardial remodeling and disturbance of cardiac function supposedly through inhibition of TGF β1 signaling. 78,164 PKG is involved in BNP-induced inhibition of TGF β1 signaling. 164 Investigators found that BNP inhibits pro-fibrotic effect TGF β1 in human primary cardiac fibroblast. 165 TGF-β1 expression is markedly enhanced in Npr1-/- mouse hearts and suppressed in Npr1++/++ mouse hearts. 30 CNP induced downregulation of TGF-β1 in isolated perfused beating rat left atria. 157 In vitro, CD-NP (synthetic NP) reduced collagen production stimulated by TGF-β1 greater than BNP or CNP in isolated cardiac fibroblasts of patients with HF. 38
The presented data indicate that the anti-fibrotic effect of natriuretic peptides may be mediated through inhibition TGF-β1 expression.
Natriuretic Peptides and Transcription Factors
It is known that transcription factors are involved in the cardioprotective effect of pre- and postconditioning. 105,151 This is primarily signal transducer and activator of transcription (STAT) and nuclear factor kappa B (NFκB). 151 There are no evince on involvement STAT in the infarct-reducing effect of natriuretic peptides. However, it was established that NFκB is involved in some cardiovascular effects of natriuretic peptides. Human umbilical vein endothelial cells (HUVEC) were pretreated with ANP before using TNF-α. 166 ANP reduced TNF-α-induced NF-κB activity and decreased translocation of p65 (one of NF-κB proteins) to nuclei. 166 It was established that TNF-α expression is enhanced by 8-fold in Npr1-/- mouse hearts compared with wild-type mouse hearts. 30 Myocardial fibrosis, total collagen, were increased in Npr1-/- mice compared wild-type animals. NF-κB activity in mouse ventricular tissues was enhanced by 4-fold and increased translocation of the p65 subunit from the cytoplasm to nuclei in Npr1-/- mice. CNP attenuated LPS-induced endothelial activation by inhibiting the NF-κB binding activity and eliminated LPS-induced intracellular ROS in HUVEC. 97 NF-κB binding activity was markedly decreased in Npr1++/++ mouse hearts compared with wild-type mice. 30
GATA transcription factors are a family of transcription factors that interact with GATA sequence in DNA and involved in the regulation of inflammation. 167 Pretreatment with GATA4 siRNA completely abolished the ability of ANP to inhibit endothelin-1 synthesis by isolated cardiac fibroblasts. 143
Thus, it has been shown that natriuretic peptides can inhibit NF-κB activity and suppress endothelin-1 synthesis through GATA activation.
Conclusion
The presented data indicate that natriuretic peptides have the infarct-limiting effect. Natriuretic peptides prevent reperfusion cardiac injury, prevent adverse post-infarction remodeling of the heart and increase survival of rats with experimental myocardial infarction. Atrial natriuretic peptide does not have the infarct-reducing effect in rats with alloxan-induced DM. Natriuretic peptides have the anti-apoptotic and anti-inflammatory effects. However, it is unclear to what extent their anti-inflammatory effect is involved in their infarction-sparing and antifibrotic effects. There is indirect evidence that natriuretic peptides inhibit pyroptosis and autophagy. Presented data indicate that natriuretic peptides inhibit ROS production in cardiomyocytes, aorta, heart, kidney and VSMC. Published data showed that natriuretic peptides can suppress aldosterone, angiotensin II, endothelin-1 synthesize and secretion and inhibit the effects aldosterone, angiotensin II on the post-receptor level through intracellular signaling events. Presented data indicate that natriuretic peptides activate guanylyl cyclase, activate PKG and PKA, and reduce the PDE-3 activity. However, it is unclear which of these changes provide increased cardiac tolerance to ischemia/reperfusion. NOS and soluble guanylyl cyclase are involved in the cardioprotective effect of natriuretic peptides. The cardioprotective effect of natriuretic peptides is mediated via activation of AMPK, PKC, PI3 K, MEK/ERK (ERK1/2), p70s6 k, Akt and inhibition of GSK-3β.The cardioprotective effect of natriuretic peptides is mediated via sarcKATP channel and mitoKATP channel opening. The cardioprotective effect of BNP is mediated via MPT pore closing. The anti-fibrotic effect of natriuretic peptides may be mediated through inhibition TGF-β1 expression. Natriuretic peptides can inhibit NF-κB activity and activate GATA. HO-1 and PPARγ may be involved in the infarct-reducing effect of natriuretic peptides.
Natriuretic peptides have the infarct-limiting effect in patients with STEMI. Natriuretic peptides prevent postinfarction remodeling of the heart. To finally resolve the question of the feasibility of using natriuretic peptides in AMI, a multicenter, randomized, blind, placebo-controlled study is needed to assess the effect of natriuretic peptides on the mortality of patients after AMI. We suggest that the use of cheap synthetic long-acting NPR agonists, which can be used in AMI, post-infarction remodeling of the heart, and cardiosurgical interventions using cardiopulmonary bypass, will open in a new era in the clinical use of natriuretic peptides.
Footnotes
Authors’ Note
This article was prepared with the support of a grant from the Russian Science Foundation16-15-10001. Section dedicated to kinases is framed within the framework of state assignments AAAA-A15-115120910024-0. Andrey V. Krylatov, Sergey Y. Tsibulnikova, Alexander V. Mukhomedzyanov, Alla A. Boshchenko, Victor E. Goldberg, Amteshwar S. Jaggi, Reinhold G. Erben contributed to acquisition, analysis, or interpretation and contributed to conception or design. Leonid N. Maslov contributed to acquisition, analysis, or interpretation; drafted the manuscript; and agreed to be accountable for all aspects of work ensuring integrity and accuracy. Attila Kiss contributed to acquisition, analysis, or interpretation; contributed to conception or design; drafted the manuscript; critically revised the manuscript; and agreed to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The article was prepared with the support of the Russian Science Foundation grant 16-15-10001.