
Abstract
Since the discovery of ischemic preconditioning (IPC) 26 years ago, numerous studies attempted to determine the mechanism of this powerful form of cardioprotection. One of the first proposed pathways of IPC suggested that the preconditioning stimulus activated phospholipase C via G-protein, and diacylglycerol released from phospholipid moieties activated protein kinase C (PKC) by translocating it from the cytosol to the sarcolemmal membranes. The major protective isoform of PKC was found to be the PKC-∊. Despite some contradictions and controversies, today even the most skeptical opponents acknowledge that PKC plays a significant role in the mechanism of IPC. During recent years, both the role and the place of PKC-∊ in the mechanism of IPC have been revised. The current review presents the evolution of the “PKC theory” and summarizes the most recent data regarding the role of PKC in IPC. In addition to classical IPC, PKC appears to play a role in the mechanisms of newer conditioning protocols, that is, remote IPC and ischemic postconditioning.
Introduction
In 1986, Murry et al described the phenomenon of ischemic preconditioning (IPC) and demonstrated that in the canine ischemia model several episodes of brief intermittent ischemia/reperfusion render the heart resistant to a prolonged episode of sustained ischemia 1 (Figure 1). This phenomenon was confirmed in rat, pig, and rabbit heart ischemia models. 2–5 The discovery of IPC triggered numerous studies aimed at determining the mechanisms of this powerful cardioprotection. Early studies identified adenosine, bradykinin, and opioids as endogenous triggers of IPC. 6–9 Subsequently, Ytrehus et al presented data implicating protein kinase C (PKC) as a major player in the mechanism of IPC, serving to transduce the protective signal elicited by the triggers of IPC. 10 The PKC transfers terminal phosphate from adenosine triphosphate (ATP) to serine and/or threonine residues in protein substrates and affects the most essential cellular functions, that is, growth, differentiation, and immediate regulatory responses. 11,12 Nineteen years since this seminal publication, the role of PKC in preconditioning has been intensively studied, and today even the most skeptical opponents acknowledge that PKC plays a significant role in the mechanism of IPC in at least some models and species. However, both its place in the signaling cascade and the consensus regarding its specific contribution to IPC have been revised. 13–18 In the current article, we will review the evolution of the concept that PKC plays a pivotal role in IPC and summarize the most recent data on its significance in the broader framework of conditioning-induced cardioprotection.
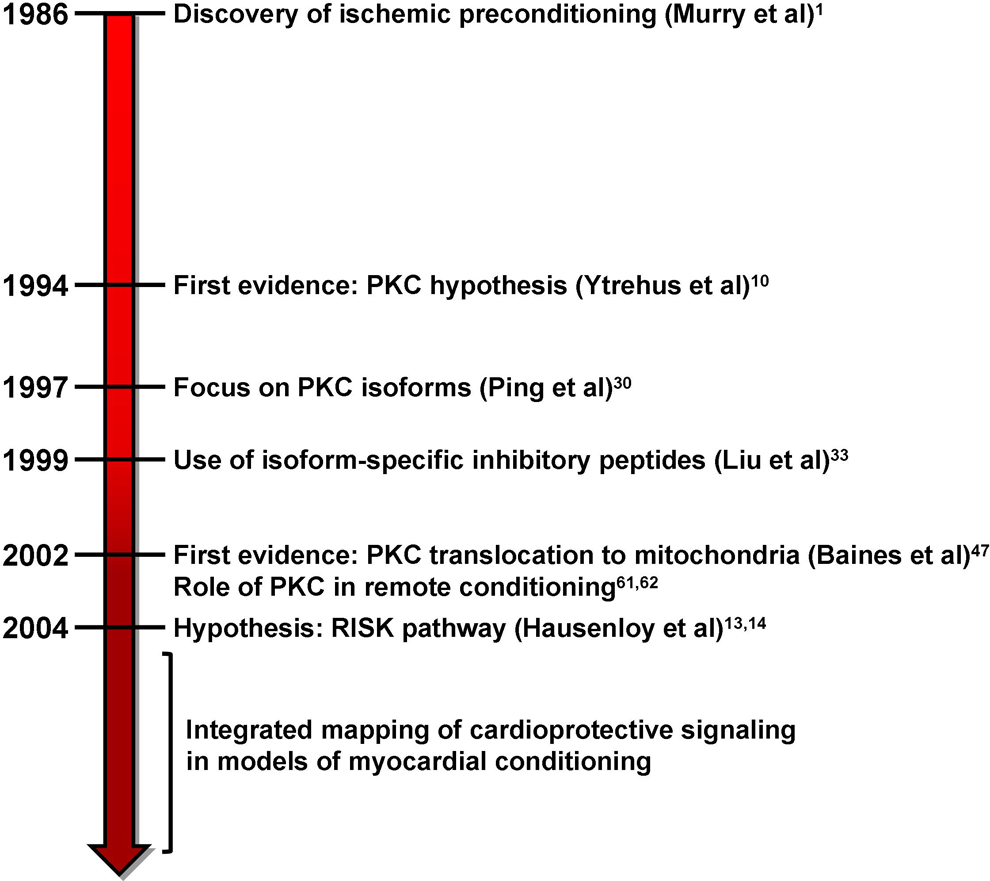
Critical milestones in the discovery and evolution of the protein kinase C (PKC) hypothesis.
Role of PKC in IPC
Genesis of the Concept
Figure 1 shows the time line of seminal advances in understanding PKC’s role in cardioprotection. The first evidence that PKC mediates the protective effect of IPC was presented by Ytrehus et al 10 (Figure 1). In their study, PKC inhibitors (staurosporine and polymyxin B), administered 5 minutes before the 30-minute sustained ischemia, abolished the protective effect of IPC in the in vivo rabbit heart ischemia model. In addition, PKC activators 4β-phorbol-12-myristate-13-acetate (PMA) and 1-oleyl-2-acetyl glycerol administered into the perfusion line of the isolated rabbit heart before the onset of sustained ischemia significantly decreased infarct size, that is, mimicked the protection rendered by IPC. 10 These data provided the impetus for the development of the “PKC hypothesis” of IPC, that is, a factor produced and/or released during brief intermittent episodes of ischemia/reperfusion (eg, adenosine) presumably stimulated receptor-associated phospholipase C, which cleaved inositol-1,4,5-trisphosphate and diacylglycerol (DAG) moieties from the membrane phospholipids. It was suggested that DAG, a hydrophobic factor, binds to PKC causing its translocation from the cytosol to cardiomyocyte membranes eliciting a yet-to-be identified protective signaling cascade. 19,20
Controversies and Contradictions
In apparent contrast to the initial data by Ytrehus et al, 10 subsequent PKC inhibitor-based pharmacological studies showed controversial results regarding the involvement of PKC in IPC. In almost every model of experimental ischemia (ie, rabbit, rat, dog, and swine), contradictory data were obtained. For example, the first study by Downey’s group demonstrated that PKC inhibitors given during the intervening brief period of reperfusion abolished the protective effect of 1 cycle of preconditioning ischemia/reperfusion. 10 However, Sandhu et al reported that PKC inhibitors had no effect on the protective effect of IPC elicited in the rabbit heart by 3 cycles of ischemia/reperfusion. 21 Multiple studies in the in vivo rat ischemia model were largely in support of the PKC role in IPC, 22–24 while others utilizing the in vitro isolated perfused rat heart ischemia model yielded opposite results and demonstrated that PKC inhibitors had no effect on preconditioning-induced protection. 25 Similar contradictory data were obtained in the in vivo canine model, 26,27 while, in the pig, preconditioning ischemia reportedly triggered PKC activation, but the use of PKC inhibitors failed to abolish its cardioprotective effect. 28,29
New Insights From Direct Assessment of PKC Translocation?
The skepticism regarding the “PKC hypothesis” of IPC grew even more when attempts to directly measure the PKC translocation in the in vivo models of experimental ischemia also provided inconclusive results. In our laboratory, we failed to demonstrate PKC translocation from the cytosol to the membrane fraction of preconditioned hearts, both in the dog and in the rabbit heart ischemia models. In these experiments, we assayed the total PKC in the cytosol and the membrane fractions and quantified its amount by measuring terminal 32 P incorporation from radiolabeled ATP into PKC-specific substrate. 27,30
Comparison of studies that supported and contradicted the PKC theory revealed that the extremely high level of diversity among models and protocols, as well as variability in the timing and dosing of PKC inhibitors, made it difficult to unanimously reject or accept the hypothesis. In addition, lack of PKC isoenzyme-specific biochemical techniques significantly hampered the attempts to prove that PKC is translocated to the cardiomyocyte membranes following preconditioning stimulus.
Introduction of PKC isoform-specific antibodies facilitated the investigation of the translocation component of the hypothesis. Using PKC isoform-specific antibodies and immunofluorescence microscopy, Banerjee et al reported the presence of PKC-α, β1, δ, ∊, η, and ζ in the whole rat heart preparation and reported PKC-∊ translocation from the cytosol to sarcolemma following the application of preconditioning stimuli (2-minute brief transient ischemia and/or treatment with bradykinin). 31 One of the most comprehensive studies was presented by Ping et al who employed Western blots to study the expression and subcellular localization of 11 PKC isoforms in the hearts of conscious rabbits (Figure 1). They found that IPC (1, 3, or 6 cycles of 4-minute occlusion followed by 4 minutes of reperfusion) did not cause significant changes in the total subcellular distribution of PKC, thus, confirming the results obtained in our laboratory. However, use of PKC isoform-specific antibodies revealed that IPC was associated with a significant translocation of PKC-∊ and PKC-η from cytosol into the particulate fraction. 32 Yoshida et al also employed the Western blot technique to study PKC translocation in the isolated perfused rat heart ischemia model. They found that 3 PKC isoforms (ie, PKC-α, δ, and ∊) translocated into the membrane fraction following 3 cycles or 3-minutes ischemia separated by 5 minutes of reperfusion, a preconditioning protocol that was confirmed to be protective (ie, improved the recovery of left ventricular-developed pressure following relief of sustained ischemia). 33 These studies underscored the importance of focusing on individual PKC isoforms rather than subcellular redistribution of total PKC, established that, during IPC, 1 or more of the PKC isoforms translocate into the cardiomyocyte membranes and thus confirmed a crucial postulate of the PKC theory—that is, PKC translocation following the IPC stimulus.
Honing the Hypothesis: Role of Individual PKC Isoforms
The next significant step in understanding the role of PKC in IPC was related to the characterization of the divergent roles of individual PKC isoforms in cardioprotection. The family of PKC includes conventional (α, β, and γ), novel (δ, ∊, η, and θ), and atypical (ζ, ι, and λ) isoforms. With regard to IPC, attention has focused largely on 2 novel PKC isoforms, that is, PKC-∊ and -δ.
The PKC-∊: A Story of Consensus
As described previously, Banerjee et al and Ping et al used isoform-specific antibodies to provide the first evidence implicating the involvement of PKC-∊ in IPC. 31,32 Corroborating data were obtained in experiments using targeted disruption of PKC-∊ gene. In this study, isolated perfused hearts from knockout mice (PKC-∊ −/−) failed to respond to IPC induced by 4 cycles of 4-minute ischemia followed by 6 minutes of reperfusion. Infarct size caused by 45 minutes of global ischemia and 90 minutes of reperfusion was significantly larger in the PKC-∊ (−/−) compared to their heterozygous (PKC-∊ +/−) counterparts. 34
A novel approach for testing the role of PKC in IPC was made available by the introduction of PKC isozyme-selective compounds (Figure 1). This approach is based on a molecular mechanism that involves highly specific anchoring of PKC isoforms to their receptors (receptors for activated C-kinase [RACKs]). This methodology allowed for the development of PKC isoform-specific inhibitory peptides that represent the RACK-binding site and prevent PKC isoform binding to its receptor. In addition, pseudo-RACK peptides were also developed, which, in contrast to the inhibitory peptides, induce selective translocation of individual PKC isoforms (for complete review see Budas et al 13 ). To test this approach, Liu et al applied IPC in isolated adult rat ventricular cardiomyocytes by pelleting for 10 minutes (designed to mimic brief ischemia) followed by resuspension in oxygenated medium. Sustained ischemia was simulated by maintaining the cardiomyocyte pellet in hypoxic buffer for 180 minutes. The resulting injury was assessed as cardiomyocyte osmotic fragility after incubation in hypotonic solution with Trypan blue. Using inhibitory peptides, they demonstrated that inhibitory peptide for PKC-∊ abolished the protective effect of IPC, while inhibitory peptides for PKCs-β, δ, and η were ineffective in attenuating IPC-induced cardioprotection. 35
The role of PKC-∊ activation in IPC was tested by Gregory et al. 36 They demonstrated that phospholamban-deficient mouse hearts exhibited significantly higher impairment with regard to the contractile recovery during reperfusion following 40 minutes of global ischemia. In addition to phospholamban deficiency, these hearts showed a decreased PKC-∊ distribution in the particulate fraction. When phospholamban-deficient mice were mated with mice expressing PKC-∊-translocation activator peptide (ie, ψ∊RACK), the resulting transgenes demonstrated increased PKC-∊ translocation into particulate fraction and improved contractility during reperfusion. 36
Taken together, the aforementioned data support the hypothesis that PKC-∊ translocation is a major step in the mechanism of IPC. Moreover, these reports, together with later studies utilizing PKC isoform-specific antibodies, manipulations with PKC-∊ gene and inhibitory, and activating PKC-∊ peptides, have resulted in widespread acceptance of the protective role of PKC-∊ in IPC. There is, however, a potentially important caveat to this consensus; the argument regarding the role of PKC-∊ in preconditioning is based on the data obtained in rat, mouse, and rabbit models; the historical controversies regarding the contribution of PKC in the canine and porcine models have not been systematically revisited using the more technically advanced isoform-specific strategies.
The PKC-δ: From Controversy to Promise to Disappointment
The consensus among studies regarding the role of PKC-∊ in IPC did not extend to other PKC isoforms, including, most notably, the δ-isoform. In 1998, Kawamura et al presented data in support of a protective role for PKC-δ in IPC. 37 Using different PKC inhibitors and Western blot technique, they reported that translocation of both PKC-δ and PKC-∊ independently contributes to the protection rendered by 3 episodes of intermittent ischemia/reperfusion in the isolated perfused globally ischemic rat heart. Four additional pieces of evidence supported an association between subcellular redistribution of PKC-δ and cardioprotection. First, using the same isolated Langendorff perfused rat heart ischemia model, Wang et al demonstrated PKC-δ translocation to mitochondria in diazoxide-induced cardioprotection. 38 Second, a protective role of PKC-δ was demonstrated in rat neonatal cardiomyocytes expressing constitutively active PKC-δ and subjected to simulated ischemia. 39 In addition, Kolar et al reported that hypoxia-induced preconditioning in rats was associated with increased amounts of PKC-δ within the cardiac particulate fraction, while Mayr et al found that preconditioning exacerbated myocardial injury in PKC-δ-deficient mice (PKC-δ −/−). 40,41
Enthusiasm regarding the protective role of PKC-δ in IPC was not, however, shared by all researchers. A study from Gross’s laboratory reported that PKC-δ was not involved in the infarct-sparing effect afforded by 1 episode of 5-minute preconditioning ischemia followed by 5-minute reperfusion in the in vivo rat heart ischemia model. 42 Accordingly, it is possible that PKC-δ contributes to IPC in the isolated rat heart ischemia model, in the isolated neonatal rat cardiomyocytes, and in the cardioprotection rendered by intermittent general rat hypoxia, but its involvement in the in vivo rat model of regional ischemia has been challenged. 37–42
Compelling evidence against the involvement of PKC-δ in cardioprotection came from a recent study demonstrating that a peptide inhibitor of PKC-δ improved (rather than attenuated) the functional recovery of human heart atrial trabeculae subjected to simulated ischemia, while, as might be expected, a peptide activator of PKC-∊ augmented their contractility. 43 In addition, Churchill et al demonstrated that IPC in the isolated perfused rat heart model caused a decrease in PKC-δ in the mitochondrial fraction, while PKC-∊ was increased. 44 These recent studies indicated that PKC-δ does not play a protective role in the heart and helped to underscore the divergent roles of PKC-∊ and PKC-δ in cardioprotection.
Based on the concept that translocation of PKC-δ may be deleterious (ie, in contrast to PKC-∊, not protective), an attempt was made to develop a cardioprotective pharmacological strategy based on the PKC-δ selective inhibition. The PKC-δ inhibitor peptide (δV1-1) was synthesized and conjugated with a carrier peptide. Intracoronary infusion of the conjugated PKC-δ inhibitor at the time of reperfusion significantly decreased infarct size and improved cardiac function in pigs subjected to 30 minutes of coronary artery occlusion. 45 However, subsequent clinical trials with this PKC-δ inhibitor peptide (delcasertib) yielded disappointing results; intracoronary administration of delcasertib prior to percutaneous coronary intervention in patients with acute ST-segment elevation myocardial infarction failed to attenuate myocardial injury and/or improve clinical outcomes. 46 This negative clinical trial will undoubtedly jeopardize future efforts to develop a new drug for acute coronary symptoms based on the PKC-δ inhibition.
The “Big Picture”: Current View on the Position of PKC in the Preconditioning-Induced Signaling Cascade
One of the first proposed schemes of IPC implied that the preconditioning stimulus activates phospholipase C via a G-protein-related mechanism, and DAG released from phospholipid moieties facilitates PKC translocation (activation) from cytosol to sarcolemmal membranes. 19 Further studies identified members of the mitogen-activated protein kinases (MAPKs) family as participants of the IPC signaling cascade. For example, Ping et al demonstrated that IPC caused significant increase in the total MAPK activity, and this increase was largely attributed to increases in p44/p42 MAPK activities. Moreover, activation of MAPK by IPC was effectively blocked by a PKC inhibitor—chelerythrine. 47 The list of prospective players involved in IPC was expanded when it was demonstrated that PKC-dependent activation of additional MAPKs, that is, p46/p54 c-Jun N-terminal kinases (JNKs) were also involved in preconditoning. 48 Finally, a landmark step toward the understanding of the exact place of individual factors involved in the preconditioning-induced signaling cascade was made by Baines et al who reported that mitochondrial PKC-∊ makes a functional signaling complex with representatives of the MAPK family, extracellular signal-regulated kinases (ERKs), JNKs, and p38. 49 Thus, for the first time, PKC-∊ as a mediator of IPC was suggested to be localized in the mitochondria (Figure 1).
A second, pivotal observation was made by Pain et al. 50 After studying mitochondrial KATP channels in the isolated rabbit heart subjected to IPC or diazoxide treatment (KATP channel opener), they proposed that IPC, via G-protein-related mechanism (1) facilitates the opening of mitochondrial KATP channels, resulting in (2) a short burst of free radical generation, and (3) subsequent activation of PKC (perhaps its ∊-isoform). Thus, for the first time, the involvement of PKC in IPC was mapped downstream of several factors that appeared to be upregulated before PKC activation and/or translocation takes place. This was a significant shift from the original PKC hypothesis, where PKC was considered to be one of the first mediators conveying a protective signal elicited by cardioprotection. 19 Further refinements regarding the role of PKC in the mechanism of IPC were made by Hausenloy and Yellon who proposed (1) the coexistence of 2 cardioprotective signaling cascades involving ERK1/2 (ie, p42/p44 MAPK) and phosphatidylinositol-3-kinase (PI3K)/Akt and notably, (2) coactivation of both kinases following relief of sustained ischemia. Considering their functional role and the timing of activation, the authors called these cascades the “Reperfusion Injury Salavage Kinase” (RISK) pathway (Figure 1). Both the components of the pathway, that is, ERK1/2 and PI3K/Akt, are thought to converge on p70S6 kinase. 15,16 The PKC was placed downstream of the RISK pathway, since it was known that PKC is activated by PI3K/Akt signaling. 51
Considering that numerous new prospective protective factors of IPC have been identified, efforts were made to devise a comprehensive scheme of classical IPC. One of the most popular schemes divides the protective signaling into 2 phases. The first response to the preconditioning stimulus, so-called “preconditioning phase,” takes place during the preconditioning stimulus. This phase includes activation of PI3K/Akt and members of the MAPK kinase family (MEK1/2, ERK1/2) via G-protein-related mechanisms, convergence of both the pathways (perhaps via p70S6 kinase) on mitochondria, generation of reactive oxygen species (ROS) with subsequent activation of PKC-∊, and opening of KATP channels. The second phase takes place during reperfusion and is aimed at decreasing reperfusion injury. During this phase, a quick burst in ROS generation can activate PKC with further phosphorylation of the participants of the RISK cascade, which in turn activate mitochondrial KATP channels and cause closure of the mitochondrial permeability transition pore. 15,16 Not all stages of both the phases are well characterized (eg, ROS generation and PKC activation during reperfusion). However, this is one of the best schemes that integrate our current knowledge regarding the protective pathways activated by IPC and the role of PKC in cardioprotection. In addition to the RISK-based pathway, some authors propose a unique place for adenosine in the signaling cascade, that is, this cardioprotective trigger may, via stimulation of A1/A3 receptors, directly activate PKC, which further acts as a mediator of IPC. 17,18 Signaling pathways involved in IPC and PKC role in cardioprotection elicited by IPC are presented in Figure 2.

Summary scheme of signaling cascades hypothetically involved in the mechanism of ischemic preconditioning. ROS, reactive oxygen species; Ado A1/A3 and Ado A2B, adenosine receptors.
Expanding the Paradigm: Role of PKC in Remote Preconditioning, Postconditioning, and Perconditioning
The IPC remains an attractive target for basic cardiologists in their efforts to translate this protective phenomenon into a clinical strategy. However, making this translation in the setting of acute coronary syndromes has been difficult, since the onset of these syndromes is unpredictable and preconditioning is, by definition, a pretreatment. A significant step toward clinical implementation of myocardial conditioning was made when the remote form of ischemic preconditioning (RIPC) was discovered. Przyklenk et al demonstrated that in the dog ischemia model repeated brief episodes of intermittent occlusion/reperfusion of the left circumflex coronary artery resulted in a significant reduction in the infarct size caused by a sustained occlusion of left anterior descending coronary artery. 52 This discovery prompted Birnbaum et al to devise a protocol of an interorgan RIPC. Transient stenosis/reperfusion of the femoral artery in rabbits (combined with electrical stimulation of gastrocnemius muscle) significantly reduced the infarct size in the heart. 53 Later, the phenomenon of RIPC was confirmed in the rat and pig ischemia models. 54,55
In the recent years, new forms of myocardial conditioning, that is, local and remote ischemic postconditioning (PostC) and ischemic perconditioning (PerC), have been described. In the case of local ischemic PostC, coronary reperfusion is initiated in a stuttered manner (that is, interrupted with brief periods of reocclusion), 56,57 while in the case of remote ischemic PostC, brief episodes of occlusion and reperfusion are applied to a vascular bed of a remote organ (eg, kidney) prior to the onset of coronary reperfusion. 58 Finally, in the models of ischemic PerC, ischemia/reperfusion cycles are applied in the vascular bed of the remote organ (eg, lower limb artery in pigs) during the sustained coronary artery occlusion phase. 59 Some of the conditioning protocols (eg, RIPC) are under investigation in clinical settings. 60–62
Protein Kinase C and Remote Preconditioning
Since involvement of PKC-∊ was established in classical IPC, it was reasonable to assume that the same PKC isoform might be involved in the newer forms of conditioning. The RIPC applied in rats by 15 minutes of mesenteric artery occlusion followed by 15 minutes of reperfusion significantly reduced infarct size caused by 30 minutes of coronary occlusion followed by 150 minutes of reperfusion. The protective effect of RIPC was accompanied by translocation of PKC-∊ from the cytosol to the particulate compartment of the heart and was blocked by bradykinin antagonist HOE140. The protection rendered by RIPC was abolished with PKC antagonist (chelerythrine) pretreatment. 63 Similarly, the involvement of PKC in remote preconditioning was implicated in a study carried out by Weinbrenner et al. In their experiments in rats, the cardioprotective effect of remote preconditioning, triggered by 15 minutes of infrarenal aortic occlusion followed by 10 minutes of reperfusion, was abolished by pretreatment with the PKC inhibitor—chelerythrine. 64
Protein Kinase C and PostC
Zatta et al 65 reported a significant reduction in infarct size in rats that were postconditioned with three 10-second cycles of reperfusion/reocclusion, applied immediately after a 30-minute period of sustained ischemia. This effect was attenuated by administration of chelerythrine or the PKC-∊ antagonist KIEI-1 (PKC-∊ inhibitor conjugated with a carrier protein) during reperfusion. In addition, PostC was accompanied by an increase in the level of phosphorylated PKC-∊ in the total cardiac cell homogenate (an indication of PKC-∊ activation). In a rabbit model of PostC, 4 cycles of 30 seconds of coronary artery reperfusion/occlusion significantly reduced infarct size after 30 minutes of occlusion followed by 3 hours of reperfusion. Chelerythrine, administered 5 minutes before reperfusion, abolished the infarct-sparing effect of PostC, while infusion of the PKC activator PMA in lieu of PostC was cardioprotective. 66 Taken together, these data indicate the involvement of PKC in the protection rendered by local ischemic PostC.
Remote PostC (ie, 5-minute renal artery occlusion applied 1-minute before left coronary artery reperfusion) reduced myocardial infarct size caused by 30 minutes of coronary artery occlusion and 3 hours of reperfusion. 58 There is no direct evidence that PKC plays a role in this form of myocardial conditioning. However, (1) administration of an adenosine antagonist (8-sulfophenyl theophylline) 5 minutes before coronary artery reperfusion reportedly abrogates the protective effect of remote PostC, 58 and (2) multiple studies have described the involvement of PKC translocation in adenosine-mediated cardioprotection. 17,18 Logic suggests that PKC may also participate in the protection rendered by remote PostC.
Protein Kinase C and Perconditioning
As with remote PostC, there is only indirect and circumstantial evidence for a possible involvement of PKC in the infarct-sparing effect of myocardial perconditioning (PerC). The PerC protocol includes remote vascular bed ischemia (eg, lower limb artery ischemia in pigs) during the sustained coronary artery occlusion. 59 Specifically, Schmidt et al demonstrated that infarct size reduction with PerC in pig model (four 5-minute cycles of lower limb ischemia with tourniquet) was abolished by the KATP channel antagonist—glibenclamide, thus suggesting that KATP channels are involved in the mechanism of PerC. 59 Given the reported role of mitochondrial PKC-∊ in KATP channel regulation during IPC, its involvement in the mechanism of ischemic PerC seems a likely scenario.
Conclusion
The IPC was first discovered by Murry et al in 1986. 1 During the past 26 years, researchers went through various phases of optimism and pessimism in their collective efforts to elucidate the cellular mechanisms of the phenomenon. As of today, a detailed mechanistic map of IPC has been devised, and the most critical steps have been identified. Most studies suggest a role for PKC isoforms in the mechanism of IPC. In addition, early evidence has implicated PKC as a mediator of both PostC and remote conditioning. Overall, there is hope that the powerful cardioprotective phenomenon of myocardial conditioning (encompassing pre-, post-, remote-, and PerC) will translate into a successful clinical strategy, and a simple and cost-effective cardioprotective therapeutic intervention will be developed.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Gwladys and John Zurlo Charitable Foundation.