
Abstract
High-density lipoprotein (HDL)-targeting therapies, including reconstituted HDL (rHDL), are attractive agents for treating dyslipidemia and atherosclerosis, as they may increase HDL levels and enhance therapeutic activities associated with HDL, including reverse cholesterol transport (RCT). Using CSL-111, a rHDL consisting of native human apolipoprotein AI (hApoAI) and phospholipids, we characterized the acute effects of rHDL administration in C57Bl/6 mice to (i) further our understanding of the mechanism of action of rHDL, and (ii) evaluate the usefulness of the mouse as a preclinical model for HDL-targeting therapies. After a single injection of CSL-111, there was a dose- and time-dependent increase of hApoAI, human pre-β HDL, total cholesterol, and triglycerides in serum, consistent with the effects of CSL-111 in humans. However, unlike in humans, there was no measurable increase in cholesteryl esters. Evaluated ex vivo, the ATP binding cassette A1 (ABCA1)- and scavenger receptor type BI (SR-BI)-dependent cholesterol efflux capacity of serum from CSL-111-treated mice was increased compared with serum from vehicle-treated animals. Fractionation by size exclusion chromatography of lipoproteins in serum from treated mice revealed hApoAI in particles the size of endogenous HDL and slightly larger, cholesterol-enriched particles of all sizes, including sizes distinct from endogenous HDL or CSL-111 itself, and triglyceride-enriched particles the size of very-low-density lipoprotein (VLDL). These results suggest that in mouse blood CSL-111 is remodeled and generates enhanced cholesterol efflux capacity which increases mobilization of free cholesterol from peripheral tissues. Our findings complement the previous reports on CSL-111 in human participants and provide data with which to evaluate the potential utility of mouse models in mechanistic studies of HDL-targeting therapies.
Introduction
High-density lipoprotein cholesterol (HDL-C) levels inversely correlate with the incidence of cardiovascular diseases. 1 This may be related to HDL's ability to promote reverse cholesterol transport (RCT) together with lipid-independent effects. 2
Pharmacological elevation of HDL-C has met with limited success. Niacin can increase plasma HDL-C, and its use has been associated with improved outcomes, but patient compliance is limited due to side effects 3 ; cholesteryl ester transfer protein (CETP) inhibitors can also increase HDL-C levels and outcomes trials are ongoing. 4 Another approach is direct increase in plasma apolipoprotein AI (ApoAI) concentration. This approach includes the small molecule RVX-208, which stimulates ApoAI synthesis, 5 peptides that may act as ApoAI mimetics, 6 –10 and reconstituted HDL (rHDL) consisting of human ApoAI (hApoAI) and phospholipids. 11–14 These approaches have resulted in favorable effects, such as increased plasma HDL or improvement in HDL function in vitro and in preclinical species. 5 ,7,15,16 However, positive effects in animals do not always translate to the clinic. For example, RVX-208, while inducing a large increase in HDL in nonhuman primates, 5 only modestly increased circulating ApoAI and HDL in humans, and dose was limited by increased circulating liver transaminases 17 ; similarly, L4F, an ApoAI mimetic peptide with efficacy in preclinical models, 18 failed to improve HDL function in humans. 19 In the case of rHDL, while human studies indicate that administration results in increased cholesterol elimination, 20 ,21 1 preclinical study failed to show a net increase in cholesterol elimination, even in the presence of increased cholesterol mobilization from peripheral tissues to plasma. 22 These inconsistent results highlight the need for a better understanding of preclinical models and assessment of their translatability into a clinical setting.
One aspect of rHDL biology, which has not been evaluated in animals is the acute global changes in circulating lipoproteins that occur after dosing with rHDL. We therefore conducted detailed characterization of the acute pharmacodynamic effects of rHDL in mice using CSL-111, a rHDL that has been extensively characterized in vitro and which has displayed positive effects in humans.11–14,23 –25 CSL-111 is composed 23 of purified hApoAI and soybean phosphatidylcholine (PC), combined at a molar ratio of 1:150. The specific lipid composition of the soybean PC used in the generation of CSL-111 has been described by Shaw and coworkers, with linoleic acid as the major fatty acid (FA) present. 14 After reconstitution, CSL-111 preparations reveal the presence of 2 distinct fractions which differ in protein–lipid ratio and diameter, a smaller fraction with 1:100 protein–lipid ratio and 12.6 ± 2.8 nm diameter, and a larger fraction with protein–lipid ratio of 1:200 and 17.7 ± 4.2 nm diameter. 11,23 Both fractions are discoidal, as assessed by electron microscopy, and have similar disc thickness (4.8 ± 0.3 nm). 23 The composition of CSL-111 would suggest that, at least in vitro, it should act as a preferred substrate for cholesterol efflux via scavenger receptor type BI (SR-BI), which favors lipid-rich particles, but not for ATP binding cassette A1 (ABCA1), which favors lipid-poor particles. 26 The efflux capacity of CSL-111 in vitro and ex-vivo is therefore an important readout in our present studies. The results reported here will also aid in the evaluation of mice as a preclinical species in which to study rHDLs and other HDL-targeting therapies.
Methods
Animals
Male C57Bl/6 mice from Jackson Laboratories (Bar Harbor, Maine) were used in these studies. All animals were housed in a temperature- and humidity-controlled facility, with a 7:00
Reconstituted Human HDL
Reconstituted human HDL (CSL-111) was provided by CSL Behring AG (Bern, Switzerland) and prepared from human plasma as previously described. 23 The preparation was supplied as a lyophilized powder that upon reconstitution with sterile water rendered a solution containing 20 mg/mL protein in 10% sucrose. The molar ratio protein–phospholipid was 1:153. All further dilutions were performed in sterile 10% sucrose.
Study Design
CSL-111 at 50, 100, or 200 mg/kg or vehicle (sterile 10% sucrose) was administered to mice at time zero, by intravenous injection (tail vein). Groups of 3 to 6 mice were then euthanized at different time points (1, 6, and 24 hours post-dose), by CO2 asphyxiation, and blood was collected by cardiac puncture. Blood was transferred to serum separator tubes and allowed to clot for 1 hour at room temperature, prior to centrifugation (10 000 g for 10 minutes). Serum was then allocated to different analyses: cholesterol, triglycerides, hApoAI, human pre-β HDL, serum cholesterol efflux, or size-exclusion chromatography (see below for specific procedures). Serum cholesterol and triglycerides were measured by a Roche Diagnostic Modular Analytics P (Roche Diagnostics Corp., Indianapolis, Indiana). Total and free cholesterol and triglycerides in the chromatographic fractions were manually analyzed using colorimetric assays purchased from WAKO Diagnostics (WAKO Chemicals, USA, Inc, Richmond, Virginia). In selected studies, total and free serum cholesterol were analyzed by the same manual system. Cholesteryl ester levels were calculated as the difference between total and free cholesterol.
Enzyme-Linked Immunosorbent Assay Quantitation of HApoAI
Human ApoAI concentrations in samples were determined using a sandwich enzyme-linked immunosorbent assay (ELISA). Immulon high-binding plates were coated overnight with 0.5 μg/mL monoclonal mouse anti-hApoAI antibody (cat#H45625M; BioDesign, Meridian Life Science, Inc., Saco, Maine) at 4°C. Plates were then blocked with Tris-buffered saline and Tween 20 ([TBST] 150 mmol/L NaCl, 50 mmol/L Tris, pH 7.5, 0.05% Tween 20) supplemented with 1% bovine serum albumin (BSA) for 1 hour at room temperature, washed with TBST, and then incubated with diluted samples (1:100 000 for whole serum and 1:10 000 for fast protein liquid chromatography [FPLC] fractions) for 2 hours at room temperature. Plates were then washed and incubated with 0.5 μg/mL biotinylated goat anti-hApoAI polyclonal antibody (cat#11B-G2a; Academy Biomedical, Houston, Texas) for 1 hour at room temperature. After washing, the plates were subjected to the Streptavidin/Europium Dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA) detection system (Envision plate reader, model #2103; Perkin Elmer, Shelton, Connecticutt). Purified hApoAI was used as a standard.
Enzyme-Linked Immunosorbent Assay Quantitation of Human pre-β HDL
The amount of pre-β HDL in serum samples was determined by a commercially available human pre-β HDL ELISA (Daiichi Pure Chemicals, Inc, Tokyo, Japan) following the manufacturer's instructions. This kit has been demonstrated to specifically detect human pre-β HDL. 27 Sample dilution for the ELISA assay was 1:4200. Concentration of human pre-β HDL was expressed as μg hApoAI per mL serum.
Lipoprotein Analysis by Size Exclusion Chromatography
Serum lipoproteins were separated by a FPLC system using a Superose 6 HR column (Amersham Pharmacia Biotech, Piscataway, New Jersey). The column was equilibrated with phosphate-buffered saline ([PBS] pH 7.4) plus 1 mmol/L ethylenediaminetetraacetic acid (EDTA) and was run at a flow rate of 0.2 mL/min. Serum samples from each treatment group were pooled, supplemented with protease inhibitor cocktail (Sigma, St Louis, Missouri) and lipase inhibitor (paraoxan, di-ethyl-p-nitrophenyl phosphate; Sigma), filtered through a 0.65 µm microcentrifuge filter, and 100 µL samples were loaded onto the column. Fractions of 0.27 mL were collected and subjected to subsequent analyses. For fractionation of the CSL-111 compound itself, CSL-111 was diluted to 1.0 mg/mL using PBS (pH 7.4), with 100 µL of the diluted sample subsequently analyzed by FPLC following the above procedure. Fractions that gave measurable absorbance at OD280 in the FPLC column system were then analyzed for their hApoAI levels.
Ex-Vivo Serum Cholesterol Efflux Assay
ABCA1-dependent serum efflux of radiolabeled cholesterol assays were performed as previously described. 28 Briefly, cells from the murine macrophage cell line J774 were seeded in 24-well tissue culture plates, labeled with [ 3 H] cholesterol for 24 hours, washed and incubated with media containing 0.2% BSA for 18 hours to allow cellular cholesterol radiolabel to equilibrate, with or without 8-(4-chlorophenylthio)-adenosine 3':5'-cyclic monophosphate [(cpt)-cAMP]. Cells were then washed once and incubated at 37°C for 4 hours with acceptor medium containing 1% mouse serum. [ 3 H] released into the media was compared with total [ 3 H] at time zero to determine the percentage release of [ 3 H] cholesterol (fractional efflux). ABCA1-dependent efflux was derived by subtracting fractional efflux of non-cAMP-treated cells from fractional efflux of cAMP-treated cells.
SR-BI-dependent serum efflux of radiolabeled cholesterol assays were performed as previously described. 26 Briefly, Fu5AH cells were seeded in 24 well tissue culture plates, labeled with [ 3 H] cholesterol for 24 hours, washed and then incubated with media containing 0.2% BSA for 18 hours to allow cellular cholesterol radiolabel to equilibrate. Cells were then washed once and incubated at 37°C for 4 hours with acceptor medium containing 1% mouse serum. [ 3 H] released into the media was compared with total [ 3 H] at time zero to determine the percentage release of [ 3 H] cholesterol as SR-BI-dependent fractional efflux.
For in vitro efflux assays of the CSL-111 compound itself, CSL-111 was diluted using PBS (pH 7.4) to concentrations of 0, 0.2, 0.5, 1.0 mg/mL. The samples were then subjected to the above described ABCA1- and SR-BI-dependent assays with the exception that the acceptor medium contained 2% of a CSL-111 dilution rather than mouse serum.
Results
In Vitro Characterization of CSL-111
CSL-111 was evaluated in cholesterol efflux assays. When cells from the murine macrophage cell line J774 labeled with [ 3 H]-cholesterol were incubated with CSL-111 (Figure 1A), the rHDL failed to induce ABCA1-mediated cholesterol efflux, even at the highest concentration tested (1 mg/mL), indicating a lack of interaction between CSL-111 and ABCA1. Consistent with this observation, when CSL-111 was analyzed with the pre-β HDL ELISA less than 1% of the total hApoAI was found as pre-β, lipid-poor ApoAI, the preferred substrate for ABCA1-dependent cholesterol efflux (not shown). In contrast, CSL-111 caused a concentration-dependent increase in cholesterol efflux when incubated with Fu5AH cells labeled with [ 3 H] cholesterol (Figure 1B), indicating that CSL-111 had the ability to mediate SR-BI-dependent cholesterol efflux.
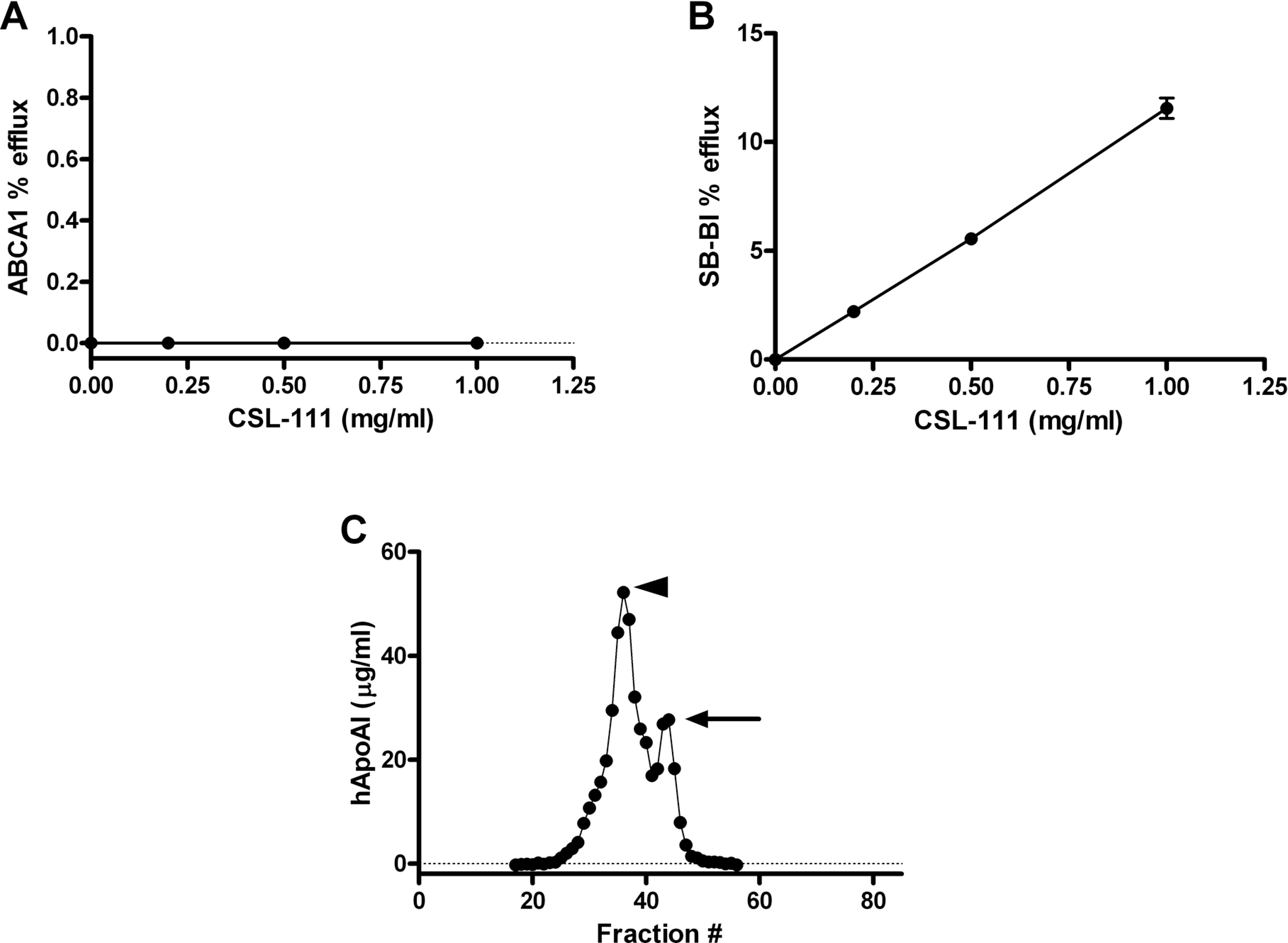
In vitro evaluation of CSL-111. A-B, ABCA1-dependent and SR-BI-dependent fractional efflux of radiolabeled cholesterol was measured in the J774 macrophage cell line and Fu5AH cell line, respectively, using the procedure described in Methods. A, CSL-111 has no effect on ABCA1-mediated cholesterol efflux in vitro. B, CSL-111 elicited dose-dependent SR-BI-mediated cholesterol efflux in vitro. C, Fractionation of CSL-111 by size exclusion chromatography indicates the presence of 2 distinct subfractions of particles, a minor fraction of HDL-sized particles (solid arrow) and a major fraction of particles larger than HDL (arrowhead). HDL indicates high-density lipoprotein.
CSL-111 was submitted to size exclusion chromatography in order to characterize its particle composition. Fractions with a positive OD280 reading were analyzed for hApoAI content by ELISA. As previously described, 23 hApoAI fractionated to 2 particle-size populations, a minor population migrating at the size expected for normal HDL particles, peaking at fraction 44, and a major population with larger particle size, peaking at fraction 36 (Figure 1C).
CSL-111 Induces Accumulation of hApoAI and pre-β HDL in Mouse Serum
Intravenous administration of CSL-111 to C57Bl/6 mice resulted in dose-dependent accumulation of hApoAI in serum (Figure 2A). Maximal levels were observed 6 hours after the highest dose (200 mg/kg) was administered. By 24 hours post-dose, hApoAI levels were below the limit of detection of our ELISA. At the 2 lower doses (50 and 100 mg/kg), maximal accumulation of hApoAI was observed 1 hour after dosing, with levels steadily decreasing over the course of the study. When the same samples were analyzed for human pre-β HDL content, using the specific Daiichi ELISA, C57Bl/6 mice dosed with CSL-111 displayed a profile of pre-β HDL accumulation similar to that observed for hApoAI, with peak levels at 6 hours post-dose for the highest dose (200 mg/kg), and peak levels at 1 hour post-dose for the lower 2 doses (Figure 2B). Pre-β HDL was not detectable in any groups at 24 hours post-dose. No hApoAI or human pre-β HDL was detectable by ELISA in vehicle-treated mice. A comparison of the levels of total hApoI and pre-β hApoAI after administration of CSL-111 to C57Bl/6 mice indicated that approximately 2% to 5% of the hApoAI resided in the pre-β fraction, which is similar to the percentage of pre-β ApoAI relative to total circulating ApoAI in normal human serum. 29
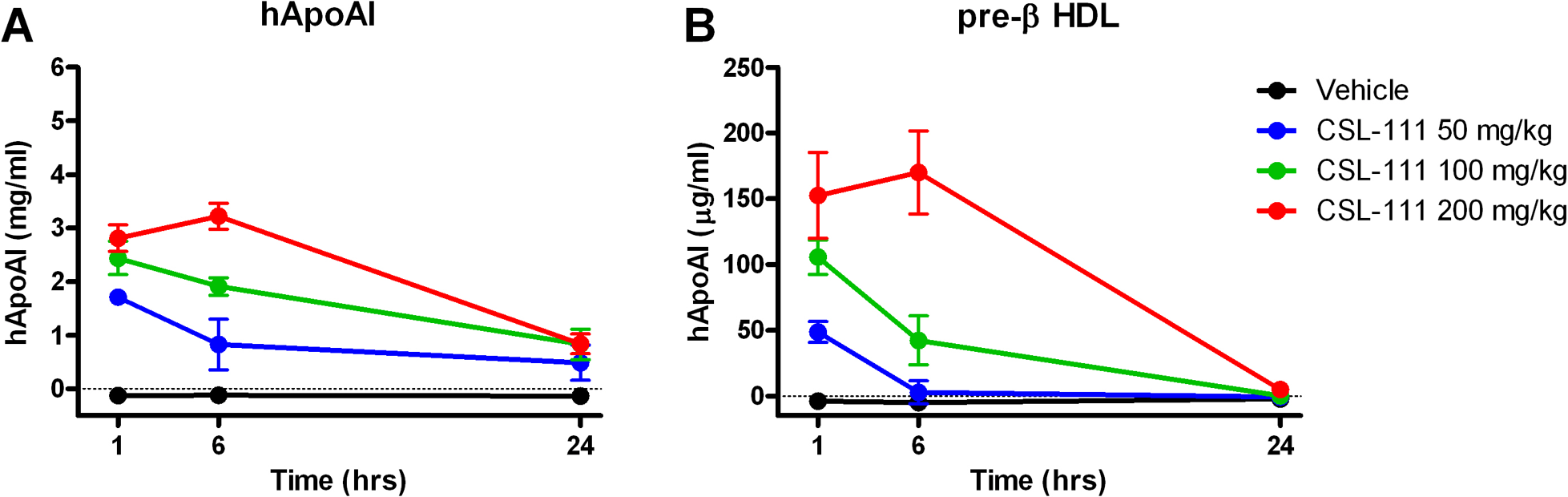
Intravenous administration of CSL-111 to mice results in time- and dose-dependent increases in hApoAI and pre-β HDL levels in mouse serum. A, C57Bl/6 mice were dosed with CSL-111 at 0 (vehicle), 50, 100, or 200 mg/kg. Six animals/dose were euthanized at 1, 6, or 24 hours post-dosing, serum collected, and hApoAI levels measured by ELISA. B, C57Bl/6 mice were dosed with CSL-111 at 0 (vehicle), 50, 100, or 200 mg/kg. Six animals/dose were euthanized at 1, 6, or 24 hours post-dosing, serum collected, and pre-β HDL levels measured by a specific ELISA. Values shown are group means ± SD. HDL indicates high-density lipoprotein; hApoAI, human apolipoprotein I; ELISA, enzyme-linked immunosorbent assay; SD, standard deviation.
Effect of CSL-111 on Mouse Serum Lipids
Intravenous administration of CSL-111 to C57Bl/6 mice resulted in time- and dose-dependent elevations of serum total cholesterol (Figure 3A). Significant elevations were observed at all doses at 1 hour post-administration of CSL-111, and cholesterol remained significantly elevated for up to 6 hours at the higher doses (100 and 200 mg/kg). At 24 hours, serum total cholesterol levels had returned to control values. Similarly, triglyceride levels were increased in C57Bl/6 mice treated with CSL-111 (Figure 3B). The kinetics of triglyceride elevation were comparable to those of total cholesterol, with significant elevations sustained for up to 6 hours at the higher doses (100 and 200 mg/kg), whereas CSL-111 at 50 mg/kg induced a significant elevation of serum triglycerides only at 1 hour. Both total cholesterol and triglyceride changes closely paralleled the exposure levels of hApoAI in these mice (compare Figure 2A with Figure 3A and B). Further analysis of the nature of serum cholesterol elevation revealed that elevations in total cholesterol were mainly driven by increases in free cholesterol, with cholesteryl ester levels remaining constant for the duration of the study at any CSL-111 dose (Figure 3C, D, and E).
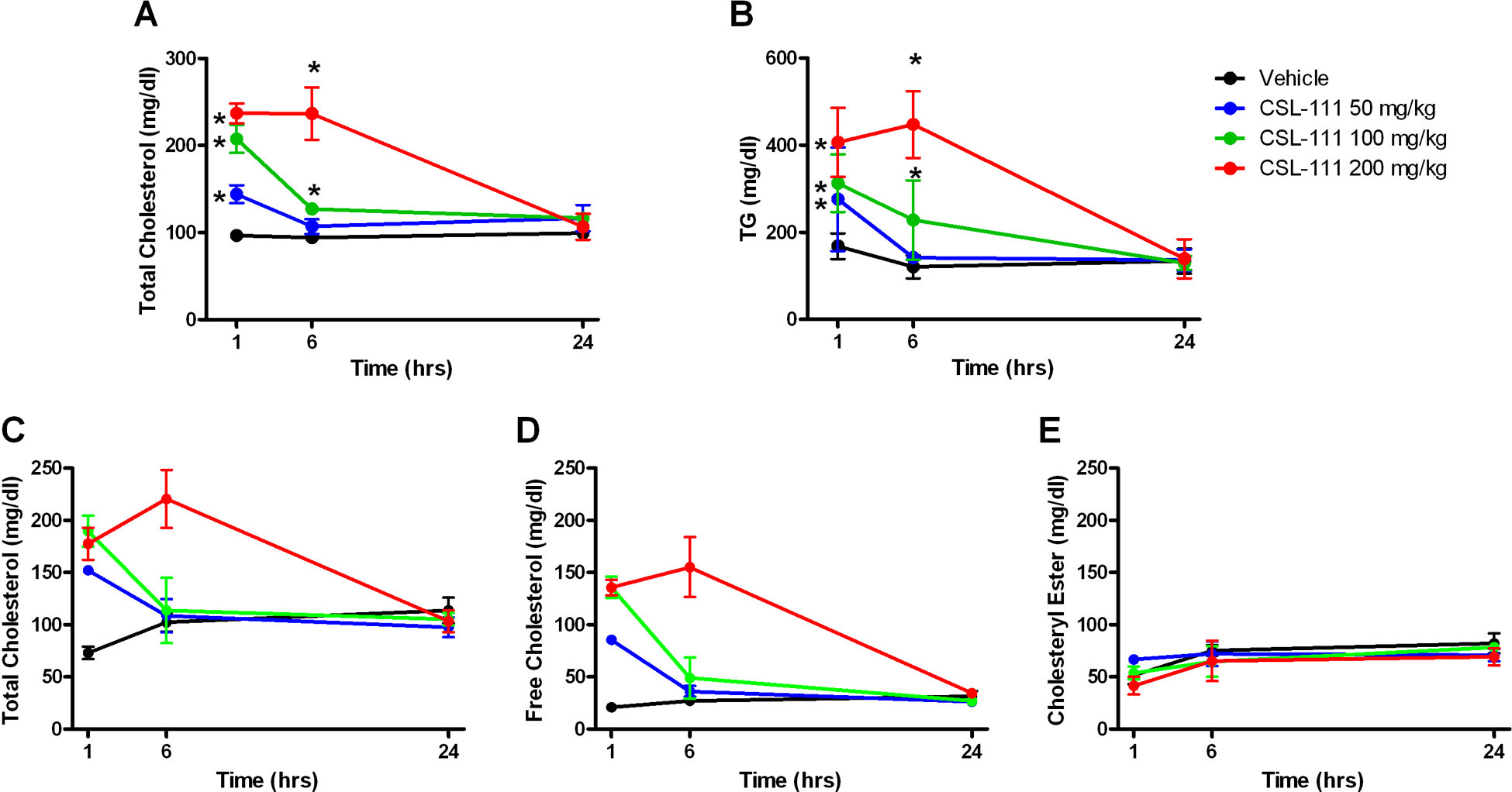
Intravenous administration of CSL-111 to mice results in time- and dose-dependent increases in total and free cholesterol and triglycerides. C57Bl/6 mice were dosed with CSL-111 at 0 (vehicle), 50, 100, or 200 mg/kg. Six animals/dose were euthanized at 1, 6, or 24 hours post-dosing, serum collected, and total cholesterol (A) or triglyceride (B) levels measured. In an independent study, serum from animals treated on the same way was analyzed for total cholesterol (C), free cholesterol (D) or cholesteryl ester (E). *P < .05 compared to vehicle group by 2-way analysis of variance (ANOVA). Values shown are group means ± standard deviation (SD).
Effect of CSL-111 on Serum Cholesterol Efflux
The serum from C57Bl/6 mice treated with increasing doses of CSL-111 for different periods of time was tested for its ability to induce efflux of radiolabeled cholesterol in a cell-based system. In contrast to what was observed with pure CSL-111 (Figure 1B and C) treatment of C57Bl/6 mice with CSL-111 resulted in a dose- and time-dependent increase in serum-dependent radiolabeled cholesterol efflux, both via ABCA1 and SR-BI (Figure 4A and B). In both instances, the profile of cholesterol efflux closely correlated with the levels of pre-β HDL and hApoAI, respectively, with r 2 = .5836 for SR-BI efflux and r 2 = .7308 for ABCA1 efflux (comparing Figures 2 and 4).
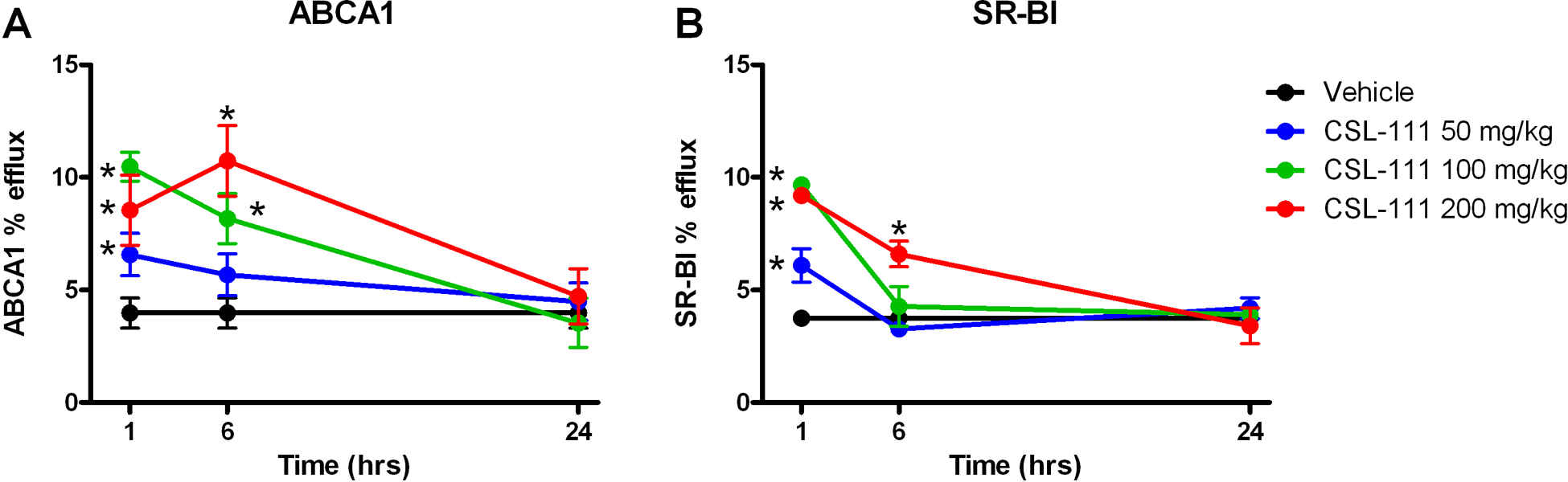
Intravenous administration of CSL-111 to mice results in time- and dose-dependent increases in SR-BI - and ABCA1-mediated cholesterol efflux. C57Bl/6 mice were dosed with CSL-111 at 0 (vehicle), 50, 100, or 200 mg/kg. Three animals/dose were euthanized at 1, 6, or 24 hours post-dosing and serum collected. ABCA1-dependent fractional efflux of radiolabeled cholesterol was measured in the murine macrophage cell line J774 (A) and SR-BI-dependent fractional efflux of radiolabeled cholesterol was measured in cell line Fu5AH (B) using the procedure described in Methods. *P < .05 compared to vehicle group by 2-way analysis of variance (ANOVA). Values shown are group means ± standard deviation (SD).
CSL-111 Induces Lipoprotein Remodeling in C57Bl/6 Mice
C57Bl/6 mice were treated with CSL-111 at 0 (vehicle group), 50, 100, or 200 mg/kg, and groups of 5 mice receiving each dose were euthanized at 1 and 6 hours post-administration of CSL-111. Serum was pooled and lipoproteins separated by size-exclusion chromatography. Human ApoAI, total cholesterol, free cholesterol, and triglycerides were measured in the collected fractions. At 1 hour post-dose hApoAI distributed to 2 distinct size populations, a smaller particle size range peaking at fraction 45, and a larger particle size range peaking at fraction 41 (Figure 5A). These fractions correspond to those which contain endogenous mouse HDL, fractions 38 to 45 (data not shown). The size distribution of hApoAI at 1 hour varied with dose: higher doses resulted in greater accumulation at smaller particle sizes. Within 1 hour of CSL-111 administration also resulted in changes in the distribution of circulating lipids (Figure 5B-D). CSL-111 induced a dose-dependent increase in total cholesterol in all fractions containing lipoprotein, from the fraction in which the smallest endogenous HDL would be found in the serum of untreated mice, fraction 45, to that which contains the largest endogenous very-low-density lipoprotein (VLDL), fraction 18 (Figure 5B). The increase in total cholesterol was mostly due to changes in free cholesterol (Figure 5C). Within an hour of treatment CSL-111 also caused a dose-dependent increase in triglycerides, localized to the VLDL fractions (Figure 5D). No changes were observed in the triglyceride content of either smaller or larger HDL-sized particles which contained hApoAI and increasing amounts of cholesterol.
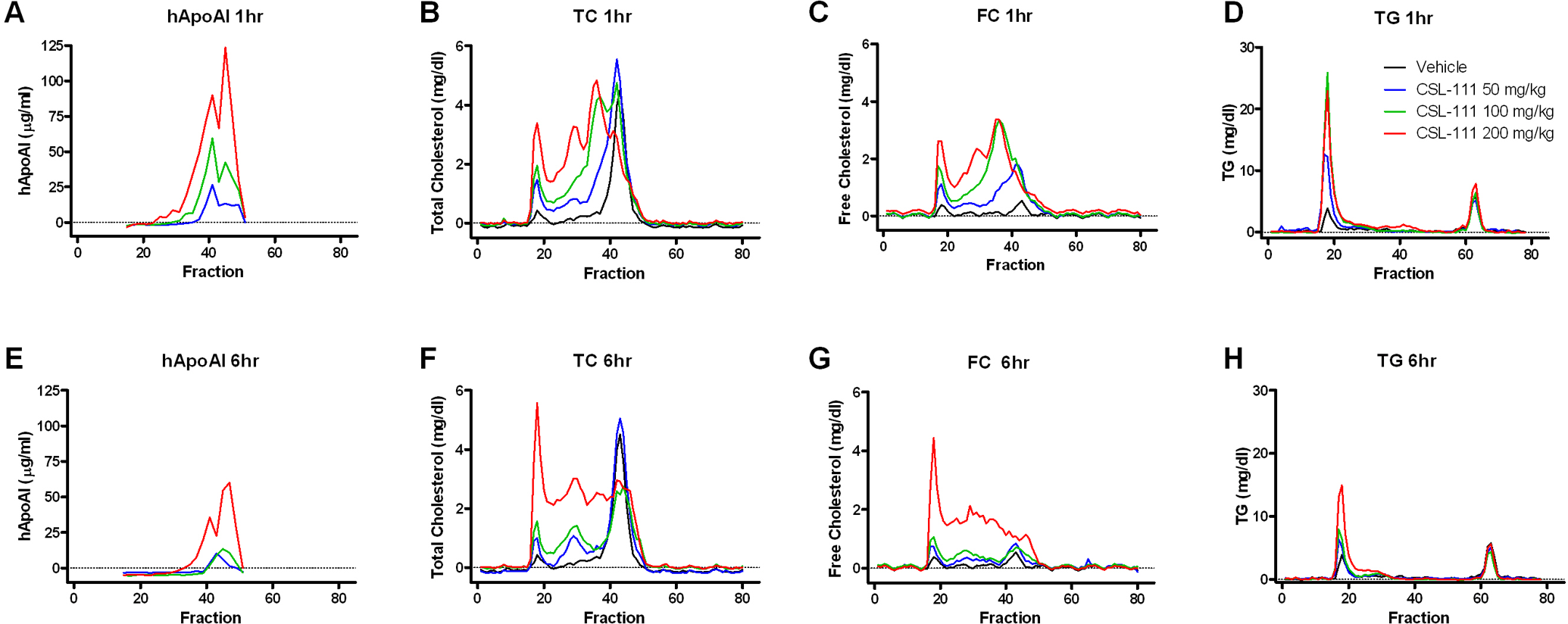
Lipoprotein profile of C57Bl/6 mice after intravenous administration of CSL-111. C57Bl/6 mice were dosed with CSL-111 at 0 (vehicle), 50, 100, or 200 mg/kg. Five animals/dose group were euthanized at 1 or 6 hours post-dosing, serum collected and pooled, and lipoproteins separated by size-exclusion chromatography. Total and free cholesterol, triglycerides, and hApoAI were measured in the indicated fractions. A-D, 1 hour post-dose. E-H, 6 hours post-dose.
The temporal response of the accumulation of hApoAI and lipids mimicked that observed when analyzing total cholesterol and triglyceride content in unfractionated serum (compare Figure 5A-D with E-H). Compared with 1 hour after treatment, cholesterol and triglyceride levels are decreasing at 6 hours in the groups receiving doses of 50 and 100 mg/kg CSL-111. In these samples, the majority of the cholesterol was associated with hApoAI-containing particles migrating at a similar size to that of normal mouse HDL (fractions 38-45), and this pattern does not change at 6 hours. In the 200 mg/kg CSL-111 group, cholesterol levels remained elevated for at least 6 hours post-administration of CSL-111 and the distribution of cholesterol shifted from a majority in hApoAI-containing particles to particles co-migrating with VLDL and a continuum of smaller particle sizes, which were mostly free of hApoA-1 (compare Figure 5A and B with E and F). As had been observed at 1 hour, the majority of the cholesterol present in these fractions was in the form of free cholesterol (Figure 5G). At 6 hours post-dose, the distribution of triglycerides did not change compared with 1 hour post-dose, remaining largely in fractions containing VLDL (Figure 5H).
Discussion
The study presented in this article was designed to gain better understanding of the pharmacodynamic effects of a rHDL, CSL-111, in mice. CSL-111 is composed of 2 populations of particles, which by electron microscopy were determined to be 12.6 nm and 17.7 nm in diameter, with protein–lipid ratios of 1:100 and 1:200, respectively. 23 Our own analysis of CSL-111 by size-exclusion chromatography confirmed the presence of 2 distinct particle populations. Consistent with its large, phospholipid-rich nature, pure CSL-111 supported only SR-BI-mediated in vitro cholesterol efflux, a process known to be favored by phospholipid-rich particles, 30 and it exhibited no effect on ABCA1-mediated in vitro cholesterol efflux, an observation consistent with the absence of pre-β HDL in the preparation. However, our data suggest that rHDL is remodeled in vivo, generating an increase in pre-β HDL-containing hApoAI, and enhancing ABCA1-dependent cholesterol efflux.
We observed that the particle distribution of hApoAI, 1 hour after dosing, was significantly different from that of pure CSL-111. The elution profile of hApoAI in CSL-111 prior to its administration to mice indicated the presence of 2 distinct peaks of hApoAI in fractions 36 (major peak) and 44 (minor peak), the latter co-migrating with the majority of endogenous HDL. In contrast, 1 hour post-dosing hApoAI appeared in 2 different locations (minor peak in fraction 41 and major peak in fraction 45), suggesting that some of the hApoAI became associated with smaller particles after injection into mice. Confirming the hypothesis that a fraction of the rHDL is remodeled to a smaller particle size, we detected dose-dependent increases in human pre-β HDL in mouse serum after administration of CSL-111, whereas we could not detect any pre-β HDL in CSL-111 itself. Consistent with these physical observations the presence of CSL-111 in mouse serum increased the ability of the serum to mediate ABCA1-dependent radiolabeled cholesterol efflux, even though CSL-111 itself has no such activity. To explain these observations, we speculate that upon delivery into mice CSL-111 is remodeled into smaller and less lipidated particles and a small percentage of the hApoAI associated with CSL-111 separates from larger particles to become the lipid-poor or lipid-free pre-β forms of HDL that interact with ABCA1.
Serum from mice dosed with CSL-111 exhibited dose-dependent increase in SR-B1-mediated cholesterol efflux. Since pure CSL-111 was also capable of mediating SR-B1-dependent cholesterol efflux in vivo, we cannot distinguish between the possibilities that the increased SR-B1 activity in serum results from activity associated with native CSL-111 itself, or with new forms of lipoprotein derived from remodeled CSL-111. However, taken together, our data strongly suggest that administration of CSL-111 to mice resulted in significant remodeling and produced lipoprotein particles that could augment RCT via the initial step of HDL formation with ABCA1 and subsequent steps of HDL formation with non-ABCA1 transporters.
Analysis of serum lipid distribution by size exclusion chromatography after dosing with 50 or 100 mg/kg of CSL-111 suggested that cholesterol rapidly associated with particles that contained hApoAI and migrated to the HDL size range. This acute increase in plasma cholesterol may reflect augmented cholesterol mobilization from peripheral tissues mediated by CSL-111 itself or by a remodeled version of CSL-111.
As the dose of CSL-111 increased and greater amounts of cholesterol accumulated in plasma, cholesterol was observed to be associated with particles spanning the size range from HDL to VLDL. Large particles newly enriched with cholesterol were devoid of measurable levels of hApoAI, suggesting that the cholesterol was accumulating on VLDL and low-density lipoprotein (LDL)-like particles. We speculate that this is due to redistribution of cholesterol removed from peripheral tissues by CSL-111-related particles to other lipoprotein particles.
CSL-111-associated cholesterol accumulation in serum was dose dependent and predominantly in the form of free cholesterol. The absolute amount of cholesteryl ester did not change over time, regardless of treatment. This phenomenon suggests that the particles mediating increased cholesterol efflux did not stimulate lecithin-cholesterol acyltransferase (LCAT) activity rapidly enough or that the efflux of cholesterol had saturated the capacity of LCAT, in the acute window in our study. Previous human data showed a different pattern.
Administration of CSL-111 to humans resulted in a transient elevation of free cholesterol, which converted into cholesteryl ester over the course of several hours, with a net increase in circulating cholesteryl ester. 11 Additional data showed that samples taken from participants infused with CSL-111 showed a transient elevation in cholesterol esterification rate ex vivo 21 and that CSL-111 can serve as a substrate for human LCAT in vitro. 23
Two factors may account for the apparent difference in cholesterol esterification in mice versus man. Francone et al 31 showed that while hApoA-I can stimulate the activity of human LCAT, it has only weak ability to stimulate murine LCAT. Species incompatibility may thus explain part of the difference. It is also known that mouse LCAT, unlike human LCAT, favors highly polyunsaturated FA (mainly 20:4 and 22:6) over 18:1 or 18:2, at the sn-2 position of PCs. 32 ,33 It is also known that the phospholipid source for CSL-111 is soybean PC, 23 which is composed mainly of 18:1, 18:2, and 18:3 FAs at the sn-2 position. 34 It is therefore possible that mouse LCAT had delayed kinetics in esterifying the large amount of newly mobilized free cholesterol using a suboptimal PC substrate. These observations highlight the need for careful evaluation of animal models as reliable predictors of human responses.
In addition to changes in total cholesterol, administration of CSL-111 to C57Bl/6 mice also resulted in a significant increase in serum triglycerides, which was dependent on the dose of CSL-111 and paralleled plasma levels of hApoAI. Analysis of triglyceride content in lipoprotein fractions indicated that it was all in the VLDL fraction, with no increased levels of triglycerides observed in the HDL fraction. This result was not surprising, as it has been previously reported that ApoAI can inhibit both lipoprotein lipase and hepatic lipase in vitro. 10,35 Examples of ApoAI-targeting agents inducing transient triglyceridemia exist in multiple studies, including ApoAI mimetic peptides dosed into mice 10 and hApoAI administered to humans 11 ,25,36 and rats, 15 and RVX-208 administered to monkeys. 5 Although the exact mechanism and physiological impacts of this phenomenon are not well understood, caution needs to be taken and an optimal therapeutic window needs to be defined in clinical development of rHDL.
In summary, our results show that CSL-111 induces effects in mice similar to those observed in humans (increased serum cholesterol efflux capacity, increased mobilization of tissue cholesterol, and increased accumulation of triglycerides in serum). However, there are aspects of CSL-111 activity in humans that are not reproduced in mice. Thus, even though CSL-111 undergoes remodeling upon administration to mice, and cholesterol is mobilized, there is no increase in serum cholesteryl esters. This could be explained by differences in LCAT affinity for the substrate, but it suggests that the use of other species, perhaps with LCAT substrate preferences closer to those of the human enzyme, may be more predictive of clinical response.
Footnotes
Acknowledgments
We are grateful to Ms Sheila Erespe of Merck for her assistance with the submission of this manuscript.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: SDW is an employee of CSL Limited; all other authors are employees of Merck Sharp & Dohme Corp.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: All authors received salary from Merck Sharp & Dohme Corp., or CSL Limited (SDW).