
Abstract
Mild hypothermia (32°C-34°C) exerts a potent cardioprotection in animal models of myocardial infarction. Recently, it has been proposed that this beneficial effect is related to survival signaling. We, therefore, hypothesized that the well-known cardioprotective pathways dependent on adenosine and/or opioid receptors could be the trigger of hypothermia-induced salvage. Open-chest rabbits were accordingly exposed to 30 minutes of coronary artery occlusion (CAO) under normothermic (NT) or hypothermic ([HT] 32°C) conditions. In the latter, hypothermia was induced by total liquid ventilation with temperature-controlled perfluorocarbons in order to effect ultrafast cooling and to accurately control cardiac temperature. After 4 hours of reperfusion, infarct and no-reflow zone sizes were assessed and quantified as a percentage of the risk zone. In animals experiencing HT ischemia, the infarct size was dramatically reduced as compared to NT animals (9% ± 3% vs 55% ± 2% of the risk zone, respectively). Importantly, administration of opioid and adenosine receptor antagonists (naloxone [6 mg/kg iv] and 8-(p-sulfophenyl) theophylline [20 mg/kg iv], respectively) did not alter the infarct size or affect the cardioprotective effect of hypothermia. Doses of these 2 antagonists were appropriately chosen since they blunted infarct size reduction induced by selective opioid or adenosine receptor stimulation with morphine (0.3 mg/kg iv) or N 6-cyclopentyladenosine ([CPA] 100 μg/kg iv), respectively. Therefore, the cardioprotective effect of mild hypothermia is not triggered by either opioid or adenosine receptor activation, suggesting the involvement of other cardioprotective pathways.
Introduction
Among the numerous strategies that have been proposed to exert cardioprotection against myocardial infarction, one of the most potent is mild hypothermia (32°C-34°C). 1,2 Such temperatures can be achieved in vivo without any external support of the circulation and result in a decreased rate of high-energy phosphate and glucose utilization. 3,4 The existence of a threshold for adenosine triphosphate (ATP) preservation between 30°C and 34°C, however, suggests that the protection afforded by mild hypothermia is in part related to the effects other than energy preservation. 5 Recently, activation of survival signaling has been proposed to mediate hypothermia-induced cardioprotection. In vitro studies in chick and mice cardiomyocytes demonstrated that cooling to 25°C or 32°C, respectively, prevented reoxygenation injury through protein kinase C-e, nitric oxide synthase (NOS), 6 and Akt pathways. 7 In isolated rabbit hearts, the infarct-limiting effect of mild hypothermia (34°C) was also abolished by pharmacological inhibition of the extracellular signal-regulated kinase (ERK) pathway. 8 Mammalian target of rapamycin (mTOR) was also recently implicated in hypothermic (HT) mouse cardiomyocytes. 9 Most of these signaling pathways were already known to be involved in cell survival following ischemia-reperfusion injury. 10 However, the trigger of these pathways during hypothermia is not known.
We hypothesized that the activation of adenosine and/or opioid G protein-coupled receptors could trigger the cardioprotective effect of hypothermia. The rationale for this hypothesis is the well-known role of these autacoids during hibernation 11–13 as well as during the initiation phase of pre- and postconditioning. 14 Accordingly, we determined whether the infarct-limiting effect of hypothermia in the in situ rabbit heart could be abolished by pharmacological inhibition of adenosine or opioid receptors with 8-(p-sulfophenyl) theophylline (SPT) and naloxone (Nal), respectively. We used total liquid ventilation with temperature-controlled perfluorocarbons to induce ultrafast hypothermia. We previously showed that this strategy can effectively use the lung as a heat exchanger to quickly change the heart’s temperature while maintaining appropriate gas exchange and results in a cardioprotective effect. 15,16
Materials and Methods
The animal instrumentation and ensuing experiments were conducted in accordance with French official regulations (agreement A94-046-13) after approval by the local ethical committee.
Animal Surgery
New Zealand White rabbits (2.5-3.0 kg) were anesthetized using zolazepam, tiletamine, and pentobarbital (all 20-30 mg/kg iv). The animals were intubated and mechanically ventilated and a left thoracotomy was performed. A thermal probe was implanted in the left atrium and a suture was passed beneath a major branch of the left coronary artery. The ends of the ligature were passed through a short propylene tube to form a snare. A coronary artery occlusion (CAO) was induced by pulling the snare. Perfusion was subsequently restored by releasing the snare. The chest was then closed in layers. Throughout the protocol, an external electrocardiogram and the arterial blood pressure from a catheter inserted in an ear artery were continuously recorded. Data were digitized and analyzed using the data acquisition software HEM v3.5 (Notocord, Croissy-sur-Seine, France).
Experimental Protocol
All rabbits had a 30-minute CAO followed by 4 hours of reperfusion. They were randomly assigned to either normothermic (NT) or mild HT ischemia (Figure 1). Ten minutes before the CAO, rabbits in HT and NT groups received either iv saline, Nal (6 mg/kg), and SPT (20 mg/kg) or a combination of both antagonists. To confirm that the doses of Nal and SPT appropriately blocked their target receptors, we investigated their ability to abolish the cardioprotective effect of morphine ([Morph] 0.3 mg/kg iv) and N 6-cyclopentyladenosine ([CPA], 100 μg/kg iv), respectively.

Experimental protocols. Morph indicates morphine; Nal, naloxone; SPT, 8-(p-sulfophenyl) theophylline; CPA, N 6-cyclopentyladenosine.
In HT groups, cooling was induced by total liquid ventilation as described previously. 15–17 Briefly, rabbits were switched to liquid ventilation by filling the lungs with 10 mL/kg of perfluorocarbon (Fluorinert, 3M, Cergy-Pontoise, France) and then connecting the endotracheal tube to the liquid ventilator. The ventilator was set to a tidal volume of ~10 mL/kg body weight with a respiratory rate of 6 breaths/min. The tidal volume was adjusted to maintain blood gases within the normal ranges. The perfluorocarbon mixture was bubbled with 100% O2. Throughout ischemia, the temperature of the perfluorocarbon was adjusted between 15°C and 32°C to maintain the left atrial temperature at ~32°C. At the end of the coronary occlusion, the perfluorocarbon was removed from the lungs and conventional gas ventilation was resumed. Animals in the HT groups were rewarmed with infrared lamps and thermal pads.
Measurement of Risk Area, Infarct Size, and No-Reflow Zones
At the end of reperfusion, the chest was reopened and a 4% thioflavin S solution (1.5 mL/kg) was rapidly infused into the left atrium. Rabbits were then sacrificed using pentobarbital followed by potassium chloride. After excision, the hearts were mounted on a Langendorff apparatus and perfused retrogradely with Alcyan blue (0.5%), following re-ligation of the coronary artery. Then the heart was cut into slices. The risk zone was identified as the nonblue region. Slices were photographed in UV light to identify the region of no-reflow as the dark zone. They were then incubated in 1% triphenyltetrazolium chloride (TTC), fixed in formaldehyde, and rephotographed. Infarcted myocardium was identified as the tissue not stained by TTC. The sizes of the areas of no-reflow, infarct, and risk zone were determined by planimetry from the pictures by a single observer blinded to the identity of the animal’s group.
Statistical Analysis
Values are expressed as means ± standard error of the mean (SEM). Hemodynamic parameters at baseline, after drug administration, during CAO (at the 15th and 25th minutes), and during reperfusion (at the 1st and 4th hours) were compared using 2-way analysis of variance (ANOVA) for repeated measures, and pairwise comparisons to the control group at each time point were made using a Fisher protected least significant difference (PLSD) test. In order to avoid multiple comparisons, values were not compared between other groups or between time points. The sizes of the risk zone (% left ventricle), infarct (% risk zone), and no-reflow zone (% infarct) were compared between the 12 groups using 1-way ANOVA followed by a Fisher PLSD test. P value <.05 was considered statistically significant.
Results
One rabbit died from ventricular fibrillation in each of the control, NT-SPT + Nal, HT-SPT, and CPA groups. In all, 76 rabbits were included in the final analysis, that is, 8 rabbits in the control NT and HT groups and 6 in each of the other groups.
Body Temperature
Left atrial and rectal temperatures were not significantly different among groups at baseline (Table 1). Throughout the experimental protocol, these temperatures were also not significantly different in the NT groups (ie, NT, NT-Nal, NT-SPT, NT-Nal + SPT, Morph, Morph + Nal, CPA, and CPA + SPT groups). As expected, temperatures were decreased during CAO in all groups subjected to hypothermia (ie, HT, HT-Nal, HT-SPT, and HT-Nal + SPT groups) and were significantly less than the temperature in the NT groups.
Left Atrial and Rectal Temperatures Among the Different Groups Throughout the Experimental Protocol a

Abbreviations: HT, hypothermia with total liquid ventilation; Morph, morphine; Nal, naloxone; NT, normothermia; SPT, 8-(p-sulfophenyl) theophylline; CPA, N 6-cyclopentyladenosine; SEM, standard error of the mean.
aValues are mean ± SEM.
b P < .05 vs corresponding NT value.
Hemodynamics
Heart rate and mean blood pressure were not significantly different between groups at baseline (Table 2). In the CPA group, heart rate was significantly decreased following CPA administration and during subsequent ischemia. This decrease was attenuated by SPT coadministration in CPA + SPT. In the other groups subjected to NT ischemia (ie, NT-Nal, NT-SPT, NT-Nal + SPT, Morph, and Morph + Nal), heart rate and mean blood pressure were not significantly different compared to those experiencing control NT. In all HT groups, the heart rate was predictably decreased during the HT phase, and averaged 40% lower than in NT groups after 15 minutes of coronary occlusion. Heart rate in HT was not further reduced by Nal or SPT. Mean blood pressure was not significantly altered in HT groups compared to NT groups.
Heart Rate and Mean Blood Pressure Among the Different Groups Throughout the Experimental Protocol a

Abbreviations: HT, hypothermia with total liquid ventilation; Morph, morphine; Nal, naloxone; NT, normothermia; SPT, 8-(p-sulfophenyl) theophylline; CPA, N 6-cyclopentyladenosine; SEM, standard error of the mean.
aValues are mean ± SEM.
b P < .05 vs corresponding NT value.
Risk Area, Infarct, and No-Reflow Sizes
Risk zone size was not significantly different among the various groups (Table 3). Infarct size was dramatically decreased by HT (9% ± 3% vs 55% ± 2% of risk zone in HT and NT, respectively; Figure 2, upper panel). Administration of Nal, SPT, or their combination did not modify the infarct size in either NT or HT groups. The no-reflow zone was likewise much smaller in HT than NT groups (Figure 2, lower panel). Similar to the infarct size, the size of the no-reflow zone in both HT and NT groups was unaffected by SPT, Nal, or their combination.

Infarct and no-reflow sizes in groups subjected to normothermic (NT) or hypothermic (HT) ischemia with or without the administration of naloxone (Nal), 8-(p-sulfophenyl) theophylline (SPT) or their combination. Open and closed circles represent individual data points and mean ± standard error of the mean (SEM) of the corresponding group, respectively. * P < .05 vs NT.
Risk Zone in the Experimental Groups a
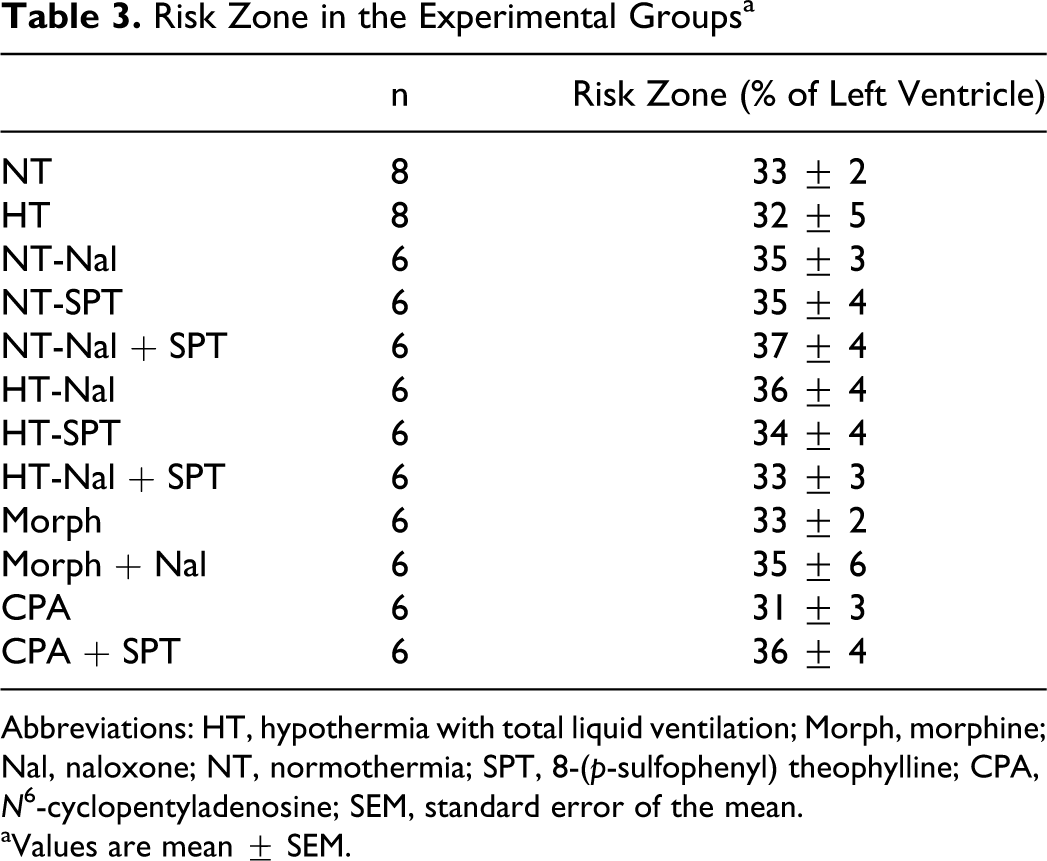
Abbreviations: HT, hypothermia with total liquid ventilation; Morph, morphine; Nal, naloxone; NT, normothermia; SPT, 8-(p-sulfophenyl) theophylline; CPA, N 6-cyclopentyladenosine; SEM, standard error of the mean.
aValues are mean ± SEM.
In order to confirm that the dosage of Nal and SPT was appropriately chosen to block their target receptors, we investigated their ability to inhibit the cardioprotective effect of Morph and CPA, respectively. Both Morph and CPA decreased infarct size compared to the control NT group; and Nal and SPT, respectively, abolished this cardioprotective effect (Figure 3, upper panel). Curiously, unlike HT which decreased both the infarct size and the no-reflow zone size, neither CPA nor Morph which decreased infarct size exerted any significant effect on no-reflow zones (Figure 3, lower panel).
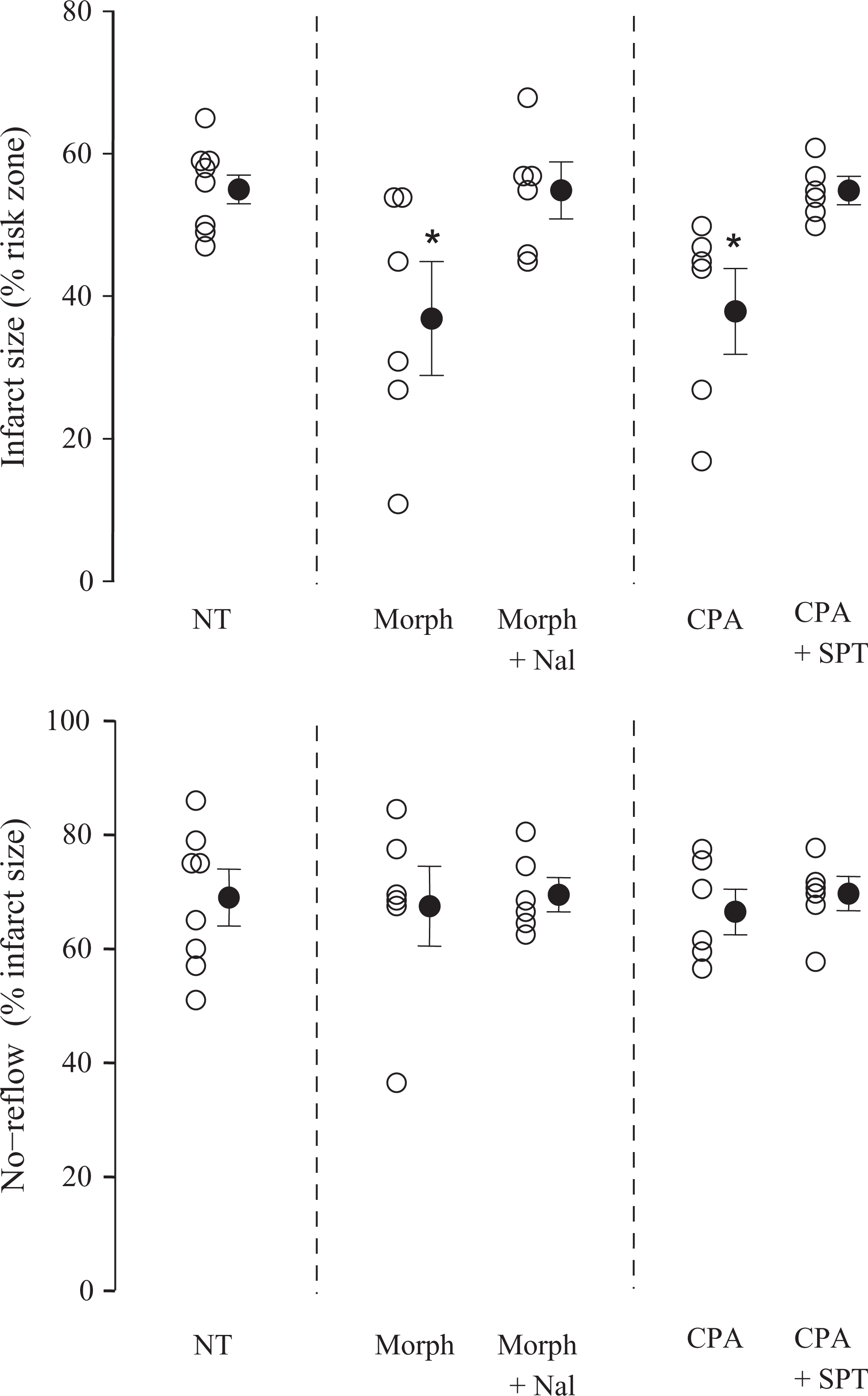
Infarct and no-reflow sizes in groups subjected to normothermic (NT) ischemia with or without the administration of morphine (Morph), N 6-cyclopentyladenosine (CPA), naloxone (Nal), 8-(p-sulfophenyl) theophylline (SPT), or their combination. The NT group is the same as that seen in Figure 2. Open and closed circles represent individual data points and mean ± standard error of the mean (SEM) of the corresponding group, respectively. * P < .05 vs NT.
Discussion
The present study demonstrates that neither adenosine nor opioid receptor blockade affected the infarct size reduction elicited by mild hypothermia in rabbits. This negative result was not related to a lack of effect of the adenosine or opioid receptor antagonists since both abolished the cardioprotection afforded by direct receptor stimulation with CPA and Morph, respectively.
The cardioprotective effect of mild hypothermia is well established in animal models of myocardial infarction. 2,18,19 The magnitude of myocardial salvage is inversely related to the length of time that transpires between the coronary occlusion and the time that the target HT temperature is achieved. 2,19 It is also proportional to the depth of hypothermia with maximal protection obtained at 32°C. 2,18 Briefly, 32°C has been the lower limit for whole body cooling because temperatures below 30°C impair the ability of the heart to maintain a normal hemodynamic state.
In the present study, we cooled the rabbits with total liquid ventilation which permitted the reduction of heart temperature to 32°C within only 2 to 5 minutes. 15,16 This confers a dramatic reduction in infarct size (-84% in HT vs NT groups). Even when HT was instituted during rather than before ischemia, its protection was greater than that afforded by pharmacological preconditioning with either 0.3 mg/kg Morph or 100 μg/kg CPA. It is unlikely that higher doses of these agonists could have provided greater protection since Morph and CPA doses were chosen on the basis of the previous studies. 20,21 One must ask whether the protective effect of HT in our study could be related to the liquid ventilation per se rather than temperature reduction. This is unlikely since we previously demonstrated in open-chest and chronically instrumented rabbits that liquid ventilation with NT perfluorocarbons does not alter infarct size. 15,16
We demonstrated that the protective effect of hypothermia is not triggered by the stimulation of adenosine or opioid receptors. Since these receptors are well known to be involved in pre- and postconditioning, 14 this demonstrates that hypothermia and adenosine- and opioid-triggered cardioprotection have distinct mechanisms despite sharing some signaling kinases. This hypothesis was previously addressed by Duncker et al. 22 In that study, they gave a cocktail of adenosine deaminase and 8-phenyltheophylline, a nonselective receptor blocker similar to the one we used, prior to coronary occlusion/reperfusion to 3 cooled open-chest pigs. They both cooled and warmed the pigs. The lowest temperature tested was 35°C in 1 animal and the infarct size was not different from that in the 2 untreated hearts cooled similarly. Two other pigs were cooled with the inhibitors. One at 36°C had an infarct size similar to that in NT hearts, while a second at 36.5°C was much more protected than 2 untreated hearts at the same temperature. We designed our study to explore much lower temperatures (32°C) and to increase our group size to achieve more statistical power.
We have recently shown in isolated rabbit hearts that ERK inhibition blocks the protective effect of mild cooling (35°C) 8 while not affecting the myocardial ATP levels. This suggests that energy preservation and ERK activation are also unrelated. Other signaling pathways have also been proposed to participate in the protective effect of hypothermia. In cardiomyocytes isolated from 1- to 2-day-old mice, 32°C hypothermia enhanced Akt/HSP27 phosphorylation and NO generation, while reactive oxygen species generation was attenuated. 7 In chick cardiomyocytes, temperature reduction to 25°C protected through increased NO generation and activation of protein kinase C-e. 6 Conversely, pharmacological inhibition of PI3K/Akt, NOS, and protein kinase C did not alter infarct size reduction with 35°C cooling limited to the ischemic period in isolated rabbit hearts. 8 A recent report also demonstrated that hypothermia activates the mammalian target of rapamycin (mTOR) in newborn mouse cardiomyocytes. 9 Possible cross talk between the different proposed pathways remains to be investigated. Our original hypothesis was that hypothermia activates survival kinases through autacoid receptor stimulation. Adenosine and opioid signaling are indeed well known to activate ERK, PI3K/Akt, NOS, and protein kinase C. 14 However, the present report shows indirectly that these autacoids are not triggering infarct size reduction during mild hypothermia. Other sarcolemmal receptors known to activate survival signaling in cardioprotection are the bradykinin receptors. 23 We did not investigate this pathway since we found no evidence of its alteration during hypothermia. Therefore, the trigger of the beneficial effect of hypothermia remains to be determined. It would have also been relevant to investigate whether hypothermia and pharmacological cardioprotection with CPA and Morph could have led to a synergistic effect. One might speculate that such an addition could occur since ischemic preconditioning and hypothermia were indeed demonstrated to afford additive infarct-size reductions. 2,24 In the present study, the great magnitude of infarct size reduction with hypothermia would have made it difficult to detect any additional protection triggered by a second intervention. Longer ischemic times and larger infarcts in HT hearts would have been necessary to test for a possible additive protective effect of CPA or Morph.
In pre- and postconditioning survival, signaling ultimately protects through inhibition of the opening of the mitochondrial permeability transition pore (MPTP). 25 We have also demonstrated that hypothermia is associated with an increased capacity of mitochondria to resist calcium before the formation of MPTP following myocardial ischemia in in vivo rabbit hearts. 16 Preserved mitochondrial biogenesis signaling was also demonstrated in isolated rabbit hearts subjected to HT cardioplegia. 26 It is not yet known whether hypothermia-induced salvage is the result of preservation of mitochondrial integrity. Profound hypothermia (<20°C) is known to alter reactive oxygen species production 27 and calcium and sodium overload. 28
The protective effect of mild hypothermia on no-reflow has been well demonstrated 17,29 and its mechanism is likely a limitation of microvascular obstruction. 30 In the present study, no-reflow was not attenuated by Morph and CPA although each reduced infarct size. Hence hypothermia likely protects both cardiomyocytes and microvessels, whereas CPA and Morph mostly protect cardiomyocytes. Importantly, the protective effect of hypothermia on no-reflow was not related to adenosine or opioid receptors. Our results also confirmed the dissociation between cardiac cell necrosis and no-reflow as previously shown. 31
In conclusion, this study demonstrates that the cardioprotective effect of mild hypothermia is not triggered by opioid or adenosine receptor activation. The signaling of hypothermia-induced cardioprotection consequently differs from that of pre- and postconditioning and further studies are required to determine the potential trigger of this protection.
Footnotes
Acknowledgment
The authors are greatly indebted to Professor J. Grassi (ITMO “Technologies pour la Santé”) and Dr C. Cans (INSERM-transfert) for their important support and advice.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Grant TLV-CARDAREST (R10028JS) from INSERM; grant ET7-460 from the “Fondation de l’Avenir”