
Abstract
Vascular endothelial growth factor (VEGF) is well known for its angiogenic activity, but recent evidence has revealed a neuroprotective action of this factor on injured or diseased neurons. In the present review, we summarize the most relevant findings that have contributed to establish a link between VEGF deficiency and neuronal degeneration. At issue, 1) mutant mice with reduced levels of VEGF show adult-onset muscle weakness and motoneuron degeneration resembling amyotrophic lateral sclerosis (ALS), 2) administration of VEGF to different animal models of motoneuron degeneration improves motor performance and ameliorates motoneuronal degeneration, and 3) there is an association between low plasmatic levels of VEGF and human ALS. Altogether, the results presented in this review highlight VEGF as an essential motoneuron neurotrophic factor endowed with promising therapeutic potential for the treatment of motoneuron disorders.
Introduction
Vascular endothelial growth factor (VEGF) was identified as the result of two parallel lines of research. In 1983, Senger and others carried out the partial purification of a protein derived from a guinea pig tumor cell line, which they named “vascular permeability factor” (VPF), due to its ability of increasing blood vessel permeability in skin. In 1989, Connolly and others isolated and sequenced, from U937 tumor cells, the human VPF molecule. In that same year, Ferrara and Henzel reported the isolation of a diffusible endothelial cell-specific mitogen from medium conditioned by bovine pituitary follicular cells, which they named VEGF to reflect the restricted target cell specificity of this molecule. The sequence of purified VEGF proved that this protein did not match any known protein in available databases (Ferrara and Henzel 1989). cDNA cloning of VEGF (Leung and others 1989) and VPF (Keck and others 1989), both reported also in 1989, demonstrated that VEGF and VPF were the same molecule. All these findings revealed that VEGF is a potent, diffusible, and specific factor for vascular endothelial cells and led to the hypothesis that this molecule could play a role in the regulation of physiologic and pathologic growth of blood vessels (Ferrara and Henzel 1989; Ferrara and others 1991; Leung and others 1989). Therefore, VEGF was historically first associated with its potent actions on endothelial cells, inducing vasculogenesis, angiogenesis, and increased vascular permeability (Apte and others 2019; Ferrara 1999, 2004; Yancopoulus and others 2000).
Subsequently, other evidence indicated that, in addition to its vascular biological activity, VEGF also acts as a neuroprotective factor. Numerous studies have demonstrated that the administration of VEGF exerts trophic effects after different types of lesions and in a variety of neuronal types. For instance, VEGF protects against seizure-induced neuronal loss in hippocampus (Nicoletti and others 2008) and attenuates status-epilepticus behavioral impairments (Nicoletti and others 2010; Ureña-Guerrero and others 2020). VEGF acts as a neurotrophic factor improving the recovery of damaged hypoxic/ischemic brain tissue (Guo and others 2016; Sun and others 2003; Yang and others 2018) and promotes the anatomic and functional recovery of injured peripheral nerves in the avascular cornea (Pan and others 2013) and after olfactory nerve bulbectomy (Beecher and others 2018). It also rescues hippocampal neurons from glutamate-induced damage (Matsuzaki and others 2001). Interestingly, VEGF exerts neurorescue effects in animal models of neurodegenerative diseases, such as Alzheimer’s and Parkinson’s, both in cerebral cortex and hippocampus (Guo and others 2019; Ureña-Guerrero and others 2020), as well as in the substantia nigra and striatum (Yasuhara and others 2005).
In addition, there is a large body of experimental evidence indicating the protective effects of VEGF in stroke. Endogenous neuronal VEGF increases in the ischemic brain and plays a protective role in the pathophysiologic processes that follow stroke (Sun and Guo 2005). The exogenous administration of VEGF reduces infarct size and decreases hypoxic neuronal death (Cárdenas-Rivera and others 2019; Sun and others 2003). Beneficial effects of VEGF include an increase in collateral vessel formation, vasodilation, angiogenesis, neuroprotection, neurogenesis, and antiapoptosis (Geiseler and Morland 2018; Guo and others 2016; Lange and others 2016).
Strikingly, although VEGF was discovered due to its angiogenic activity, this factor has an evolutionarily ancient role as a neurotrophic factor controlling neural cell morphogenesis in invertebrates. Thus, the nematode worm Caenorhabditis elegans lacks a vascular system, but VEGF is present as a trophic molecule necessary for the development of its nervous system. In insects, such as Drosophila melanogaster, which have a rudimentary vasculature, VEGF controls nervous system development. In vertebrates, which have an elaborated network of vessels, VEGF controls both nervous and vascular development, suggesting that vessels co-opted VEGF signaling from nerves to regulate their development (Zacchigna and others 2008).
VEGF Family and Receptors
The VEGF family members are dimeric glycoproteins of approximately 40 kDa. In mammals, the VEGF family consists of five members with a high degree of homology, which comprises VEGF-A (the founding molecule of this family, also known as VEGF), VEGF-B, VEGF-C, VEGF-D, and the placental growth factor (PlGF). In addition, proteins that are structurally related to the VEGFs are present in parapoxvirus (VEGF-E) and in snake venom (VEGF-F) (Olsson and others 2006). VEGF family members have been implicated in angiogenesis and lymphangiogenesis, and they may play different roles in the nervous system (e.g., neuronal wiring in the CNS, peripheral nerve development and vascularization, neural migration, neurogenesis, neurite outgrowth, neuronal survival, synaptic plasticity, neuroprotection, and neuroregeneration) (Lladó and others 2013; Ruiz de Almodovar and others 2009). They have also been implicated in the etiology and treatment of various neurologic diseases (Lange and others 2016). VEGF-B is a weak angiogenic stimulator and has a relatively restricted angiogenic activity in the ischemic heart (Li and others 2008). VEGF-B also acts as a neuroprotective agent against neurodegeneration and physiologic alterations (Calvo and others 2018; Dhondt and others 2011; Poesen and others 2008). The molecules VEGF-C and VEGF-D act on the lymphatic endothelium promoting the process of lymphangiogenesis (Lohela and others 2009). PlGF stimulates angiogenesis selectively under pathologic conditions (Carmeliet and others 2001).
The signaling of VEGFs is carried out through binding to three cell surface membrane receptors, which are endowed with tyrosin kinase intracellular activity. Each VEGF factor binds with different affinities and selectivities to these three receptors: VEGFR-1 (Flt-1), VEGFR-2 (KDR/Flk-1), and VEGFR-3 (Flt-4). VEGFRs are formed by two monomers with several extracellular immunoglobulin domains each. These mediate ligand binding. The monomers also contain a transmembrane chain and an intracellular effector comprising two regions with tyrosine kinase activity. VEGFRs form homodimers but also heterodimers of unknown function (Ferrara 2004; Ruiz de Almodovar and others 2009). VEGF binds to VEGFR-1 and VEGFR-2, VEGF-B and PlGF bind to VEGFR-1, and VEGF-C and VEGF-D interact with VEGFR-3 and VEGFR-2 (Lange and others 2016; Fig. 1).
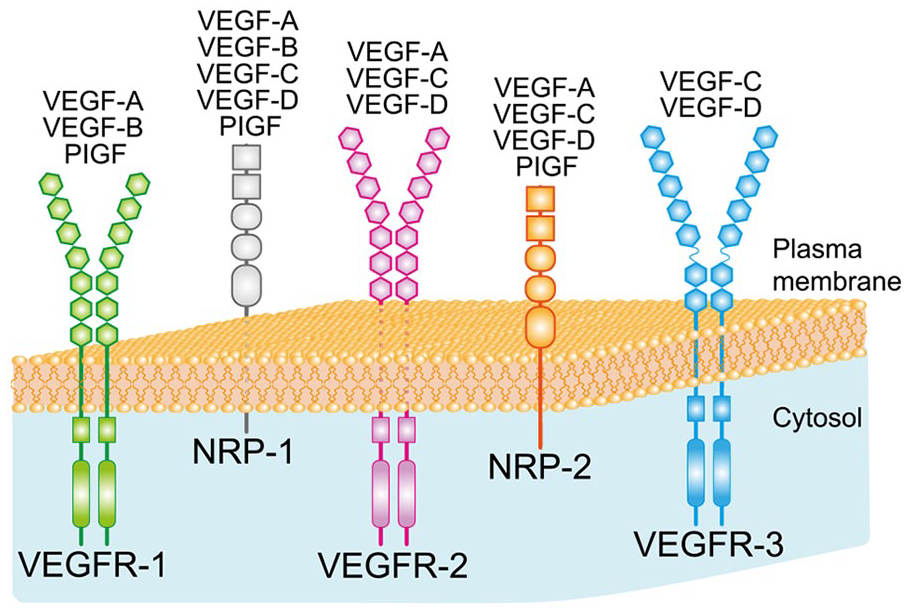
Vascular endothelial growth factor (VEGF) family and receptors. The figure is a representation of the members of the VEGF family and the specific binding of each member to the different VEGF receptors. In addition to VEGF-A (also known as VEGF), the other isoforms of the VEGF family in mammals are VEGF-B, VEGF-C, VEGF-D, and placental growth factor (PlGF). VEGF receptors include the tyrosine kinases VEGFR-1, VEGFR-2, and VEGFR-3 and the coreceptors neuropilin 1 (NRP-1) and neuropilin 2 (NRP-2).
VEGFR-2 is the best characterized signaling receptor, promoting angiogenesis in health and disease; it stimulates endothelial cell proliferation, survival, migration, and vascular permeability. VEGFR-2 also signals numerous processes in various neural types (Carmeliet and Ruiz de Almodovar 2013; Ruiz de Almodovar and others 2009). VEGFR-2 has high tyrosine kinase activity (Takahashi and Shibuya 2005). Although VEGFR-1 was discovered before VEGFR-2, its role is still unclear. VEGFR-1 has a weak tyrosine kinase activity but binds VEGF with at least 10-fold higher affinity than VEGFR-2 (Takahashi and Shibuya 2005). Consequently, signaling through VEGFR-2 promotes the phosphorylation of a larger number of intracellular effector proteins due to its strong tyrosine kinase activity. As a result, VEGFR-2 is considered the principal mediator of VEGF actions. On the other hand, due to the higher affinity of VEGFR-1 for VEGF, it has been proposed that VEGFR-1 could act as a “decoy” receptor that traps VEGF and thus prevents excessive activation of VEGFR-2 (Ferrara and Davis-Smyth 1997; Koch and Claesson-Welsh 2012). That is, VEGFR-1 would negatively regulate the activity of VEGFR-2. However, in an experimental model of stroke in rats, it has been reported that VEGFR-1 activation by VEGF promotes neuronal protection and prevents large-volume infarcts that highly correlate with neurologic performance, while the concomitant activation of VEGFR-2 reduces this effect (Cárdenas-Rivera and others 2019). The authors propose the existence of a VEGFR-2–mediated antagonism of VEGFR-1. It would be interesting to check whether this proposed antagonism between both receptors is also operative in other lesion models. The neuroprotective role of VEGFR-1 through the specific binding of VEGF-B has also been demonstrated in other systems (Calvo and others 2018; Poesen and others 2008). Therefore, further investigation is required to elucidate the role of VEGFR-1 signaling, as it might not always function as a “decoy” receptor. The third tyrosine kinase VEGFR, VEGFR-3, is present in the endothelial cells of lymphatic vessels, and it binds the ligands VEGF-C and VEGF-D (Takahashi and Shibuya 2005).
The members of the VEGF family also selectively bind two coreceptors named neuropilins (NRPs): neuropilin 1 (NRP-1) and neuropilin 2 (NRP-2) (Fig. 1). They were initially identified as semaphorin receptors, proteins involved in axonal guidance. NRPs are cell surface glycoproteins whose intracellular domains are short and lack tyrosine kinase activity. Nevertheless, when coexpressed with VEGFRs, NRPs enhance the binding to VEGFRs and increase the effectiveness of VEGFR-mediated signaling transduction (Ferrara and others 2003; Takahashi and Shibuya 2005).
The specific ligands for VEGFRs and NRPs are illustrated in Figure 1 and are as follows: VEGF (i.e., VEGF-A) binds to VEGFR-1, VEGFR-2, NRP-1, and NRP-2; VEGF-B binds to VEGFR-1 and NRP-1; PlGF binds to VEGFR-1, NRP-1, and NRP-2; and VEGF-C and VEGF-D interact with VEGFR-3, VEGFR-2, NRP-1, and NRP-2 (Ferrara and others 2003; Lange and others 2016; Takahashi and Shibuya 2005).
Regulation of VEGF by Hypoxia
Hypoxia is the principal stimulus for VEGF expression. When local oxygen tension decreases, VEGF levels rise, promoting angiogenesis (i.e., the formation of new blood vessels from preexisting ones, which counteracts the oxygen deficit in the tissue). Thus, the regulation of VEGF by hypoxia plays an adaptive, homeostatic role in the maintenance of a proper delivery of oxygen and nutrients to areas with low vascular perfusion (Pronto-Laborinho and others 2014). Some studies have shown that the hypoxia-regulated mechanisms of the VEGF gene and the erythropoietin gene share similarities (Pronto-Laborinho and others 2014).
The increase in VEGF protein levels under hypoxic conditions is due to control at both the transcriptional and translational level. At the transcriptional level, the gene sequence involved in hypoxia regulation is termed the hypoxia-response element (HRE) and has been located in the promoter of the human and murine VEGF gene (Ferrara 2004). Hypoxia-inducible transcription factors (HIFs) bind HRE and thereby up-regulate VEGF transcription under hypoxic situations. HIF is a heterodimeric protein formed by two constitutively expressed subunits: HIF-1α (120 kDa) and HIF-1β (92 kDa) (Oosthuyse and others 2001; Storkebaum and others 2004; Wang and others 1995). Elevated VEGF levels can be detected in the brain as early as 2 to 4 hours after the onset of induced focal cerebral ischemia in rodents, with a peak between 12 and 48 hours. The increase in VEGF after hypoxia lasts for at least 28 days, and it has been proposed that this VEGF increase leads to the angiogenesis that occurs after cerebral ischemia (Geiseler and Morland 2018; Marti and others 2000; Takahashi and Shibuya 2005; Zhang and others 2002).
Under normoxia (Fig. 2), HIF-1α is hydroxylated at two proline residues by the enzymatic action of proline hydroxylase proteins (PHDs), which require molecular oxygen to carry out this action. This step is necessary for the interaction with the von Hippel-Lindau (VHL) protein, a component of the E3 ubiquitin ligase complex, tagging HIF-1α for degradation (Pronto-Laborinho and others 2014; Pugh and Ratcliffe 2003; Salceda and Caro 1997). The proteasome degradation of HIF-1α precludes VEGF transcription in normoxia (Fig. 2). However, in hypoxia, PHDs are inactive due to lack of O2, and consequently, HIF-1α is not hydroxylated and is thus not ubiquitinated for proteasome degradation. Therefore, HIF-1α protein levels increase and HIF-1α translocates into the nucleus and associates with HIF-1β and the coactivators P300/CBP. This complex binds to HRE in the VEGF gene promoter, activating VEGF transcription (Semenza 2001, 2010). In this way, VEGF levels increase in response to hypoxia (Fig. 2). HIF-1α can also increase in response to excitotoxicity (Vazquez-Valls and others 2011), and the levels of VEGF consequently rise under this neurotoxic condition (Castañeda-Cabral and others 2017).
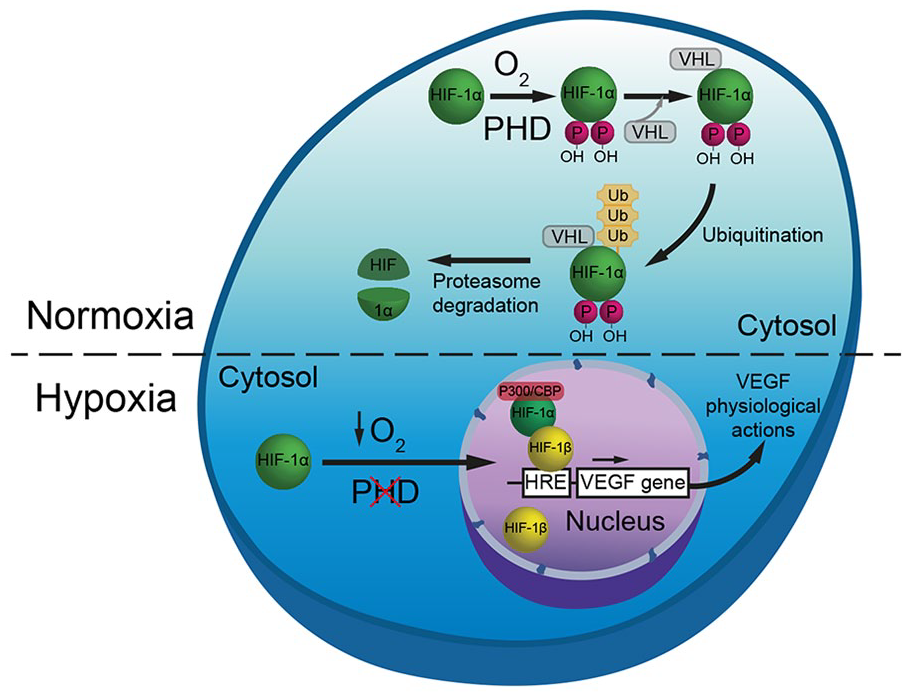
Regulation of vascular endothelial growth factor (VEGF) by hypoxia. Under normoxia, hypoxia-inducible transcription factor 1α (HIF-1α) is hydroxylated by proline (P) hydroxylase domain-containing proteins (PHDs), which require O2 for their enzymatic activity. Hydroxylation of HIF-1α allows its interaction with the von Hippel-Lindau (VHL) protein, a component of the E3 ubiquitin ligase complex. This interaction results in ubiquitination (Ub) and degradation by the proteasome pathway of HIF-1α and, consequently, prevents VEGF transcription in normoxia. Under hypoxic conditions, PHDs are inactive due to lack of O2. Therefore, HIF-1α cannot be hydroxylated and is thus not ubiquitinated for proteasome degradation. This allows HIF-1α to enter the nucleus and dimerizes with HIF-1β, and in association with the p300/CBP coactivators, this complex binds to hypoxia-response element (HRE) in the VEGF gene promoter, activating VEGF transcription.
VEGF can also be regulated at the translational level. Hypoxia promotes the stabilization of VEGF mRNA through proteins that bind to sequences located in the region UTR (3′ untranslated region) of the mRNA such as HuR (Levy and others 1998; Pronto-Laborinho and others 2014; Storkebaum and others 2004). It should be mentioned that VEGF receptors are also increased by hypoxia, as reported in endothelial cells of lung, heart, brain, kidney, and liver following systemic hypoxia (Marti and Risau, 1998), as well as in skeletal muscle in response to chronic ischemia (Milkiewicz and others 2003). The response of receptors to hypoxia enhances VEGF signaling, favoring increased vascular perfusion in ischemic tissues.
VEGF and Amyotrophic Lateral Sclerosis: Evidence from Transgenic Animals
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease characterized by rapidly progressive degeneration of motoneurons in the spinal cord, brainstem, and premotor neurons in the motor cortex, leading to severe weakness and muscle atrophy. The mean survival of patients with ALS is three to five years after symptom onset. Most ALS cases are sporadic, but approximately 10% are familial, one fifth of which are caused by mutations in the gene encoding the antioxidant Cu/Zn superoxide dismutase (SOD1). The pathogenesis of ALS appears to be a complex interplay of several processes, such as oxidative stress, excitotoxicity, protein aggregation, mitochondrial dysfunction, deleterious effects derived from glial cells, abnormal axonal transport, and deficits in neurotrophic factors, which ultimately lead to motoneuron death. However, the precise pathophysiologic mechanisms of motoneuron degeneration in ALS are still unknown. The only drug currently approved worldwide in ALS is riluzole, which acts by decreasing glutamate activity in the CNS, although its impact on survival is modest, and ALS is still an incurable disease (Bruijn and others 2004; Dhasmana and others 2022; Kaur and others 2016; Robberecht and Philips 2013; Valko and Ciesla 2019). Mice and rats expressing a mutant SOD1 transgene develop ALS with clinical, anatomic, and pathologic features that are highly reminiscent of those found in human patients with ALS. SOD1 transgenic mice and rats are a classic animal model for the study of ALS and have been widely used in numerous ALS-related investigations (Tovar-y-Romo and others 2009).
The link between VEGF and ALS was established in a fortuitous way. Attempts were made to generate knockout mice that failed to express VEGF, or either of its two principal receptors (VEGFR-1 or VEGFR-2), in studies aimed at investigating the relevance of hypoxic regulation of VEGF-dependent angiogenesis. However, these knockout mice were not viable, and they died at an embryonic stage due to lack of development of the vascular system (Greenberg and Jin 2004). To overcome this obstacle, Oosthuyse and others (2001) generated mutant mice in which the HRE sequence in the VEGF gene promoter was deleted (VEGFδ/δ mice). These genetically engineered mice resulted in an unexpected phenotype, unveiling a novel role of VEGF in the survival of adult motoneurons.
VEGFδ/δ mice exhibited impaired up-regulation of VEGF by hypoxia in neural tissue, so that spinal VEGF levels were reduced by 75%. Strikingly, these VEGFδ/δ mice developed symptoms of motoneuron disease beyond five months of age. They became progressively less mobile and showed signs of severe muscle weakness and limb paresis. Behavioral tests revealed impairment of motor coordination and muscle performance. However, their pain threshold was normal, indicating that motor, not sensory, functions were primarily affected. Electromyographic recordings showed absence of spontaneous activity in several skeletal muscles in contrast to VEGF+/+ mice. The histologic examination of muscles revealed signs of atrophy, which seemed to be due to denervation. HRE deletion caused a late-onset progressive degeneration of motoneurons, affecting the ventral horn of the spinal cord and the motor nuclei in the brainstem, with a significant loss of motoneurons and a prominent reactive astrogliosis beyond 7 months of age. The peripheral nerves of VEGFδ/δ mice progressively lost 30% of the large myelinated motor axons and showed prominent signs of Wallerian degeneration (Fig. 3A,B). Ultrastructural examination of motoneuron cytoplasm further indicated fewer Nissl bodies and abnormal mitochondria and cell organelles. At a more advanced stage of degeneration, shrunken motoneurons presented vacuolization, fewer ribosomes, and an irregular nucleus with peripheral clumping of chromatin aggregates (Fig. 3C,D). VEGFδ/δ mice did not exhibit obvious abnormalities in central structures such as hippocampus, neocortex, cerebellum, thalamus, and striatum, indicating that VEGF deficiency selectively impaired motoneurons (Carmeliet and Storkebaum 2002; Lambrechts and others 2004; Oosthuyse and others 2001; Storkebaum and others 2004).
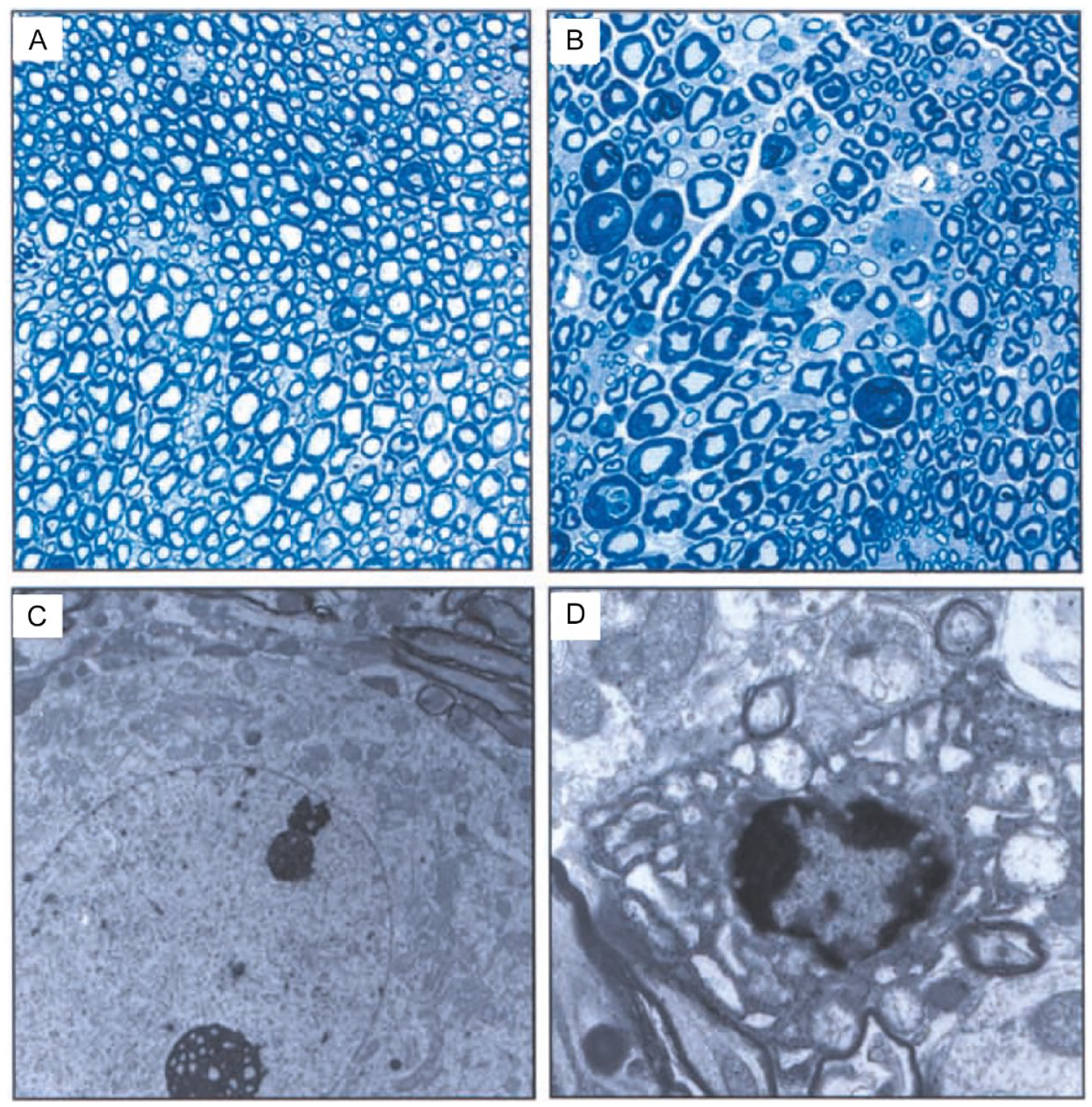
Motoneuronal degeneration in VEGFδ/δ mice. (A, B) Images of sciatic nerve semithin sections from wild-type (A) and VEGFδ/δ mice (B). In contrast to the wild type (A), the sciatic nerve of the VEGFδ/δ mice shows signs of axonal loss and Wallerian degeneration (B). (C, D) Transmission electron microscopy images of spinal cord motoneurons from wild-type (C) and VEGFδ/δ mice (D). The motoneuron in C shows typical ultrastructural characteristics of this cell type such as a large round nucleus, with homogeneous chromatin and a conspicuous nucleolus, and a cytoplasm containing a prominent rough endoplasmic reticulum and mitochondria. The ultrastructure of the VEGFδ/δ motoneuron (D) shows signs of clear degeneration. Note cell shrinkage, an irregular nucleus with aggregates of peripheral chromatin, and a reduced cytoplasm with vacuolized mitochondria and endoplasmic reticulum. VEGF, vascular endothelial growth factor. Reprinted from Carmeliet P, Storkebaum E. 2002. Vascular and neuronal effects of VEGF in the nervous system: implications for neurological disorders. Semin Cell Dev Biol 13(1):39–53. Copyright (2002), with permission from Elsevier.
All these findings indicate that VEGFδ/δ mice have severe adult-onset muscle weakness and motoneuron degeneration. The clinical symptoms and the neuropathologic alterations observed in VEGFδ/δ mice are like those seen in humans with ALS and also similar to the SOD1 mutant mice that, as mentioned above, represent a well-established model of ALS.
A molecular relationship has been found between SOD1 and VEGFδ/δ mutant mice. It has been reported that in SOD1 mutant mice, the expression of VEGF mRNA in the spinal cord declines significantly early in the course of the disease. Mutant SOD1 impairs posttranscriptional regulation of VEGF mRNA by sequestering key regulatory RNA-binding proteins (HuR and TIAR). In this way, mutant SOD1 destabilizes VEGF mRNA and down-regulates VEGF protein production. These findings led to the postulation that the resultant destabilization of VEGF mRNA critically reduces the level of this neuroprotective growth factor and accelerates the neurodegenerative process that occurs in mutant mouse model of SOD1 (Lu and others 2007, 2009).
Experiments of crossbreeding between transgenic mice have reinforced the role of VEGF as a neuroprotective factor for diseased motoneurons. Thus, crossbreeding of VEGFδ/δ with SOD1 mice produces VEGFδ/δ/SOD1 double mutants that show increased severity of motoneuron degeneration and earlier onset of muscle weakness than mice carrying the SOD1 gene alone (Lambrechts and others 2003). Similarly, when SOD1 mice are crossed with mice overexpressing VEGF, the double transgenic SOD1/VEGF+/+ mice show delayed motoneuron degeneration and motor impairment, as well as prolonged survival compared with SOD1 single transgenics (Wang and others 2007). Thus, if VEGF levels are reduced, the risk of developing ALS-like symptoms increases dramatically.
The deficiency in neurotrophic factors other than VEGF is not causal to ALS. Although the over- or underexpression of BDNF, GDNF, CNTF, or LIF, among others, affects the pre- and postnatal development of motoneurons, and although adenoviral gene transfer of some of these molecules promotes motoneuron survival in animal models of ALS, the lack of these other neurotrophic factors does not induce motoneuron degeneration or paralysis in mice, as in ALS (Lambrechts and Carmeliet 2006). However, it must be emphasized that neurotrophic factors, such as neurotrophins, GDNF, IGFs, CNTF, LIF, or CT-1, have important roles in cell survival during development, after injury and in response to disease (reviewed in Gould and Oppenheim 2011; Tovar-y-Romo and others 2014).
Effects of VEGF Administration on Neurodegeneration
VEGF delivery has been shown to ameliorate neurodegeneration. Exposure of cell cultures to the combination of hypoxia and hypoglycemia induces consistent cell death, and VEGF treatment significantly protects motoneurons from cell death (Van Den Bosch and others 2004). VEGF protects the NSC-34 motoneuron-like cell line exposed to cerebrospinal fluid from patients with ALS (Kulshreshtha and others 2011; Vijayalakshmi and others 2015). Similarly, VEGF protects NSC-34 cells transfected with adenovirus containing the mutant SOD1 protein via the PI3-K/Akt pathway responsible, among others, for cell survival, growth, and angiogenesis (Li and others 2003).
In vivo studies have demonstrated neuroprotection by VEGF. When a lentiviral vector expressing VEGF was injected in several muscles of the SOD1 mouse model of ALS, both neuroprotection and increased life expectancy occurred (Azzouz and others 2004). Intrathecal spinal cord transplantation of immortalized human–VEGF-expressing–neural stem cells delayed onset and prolonged the survival of SOD1 transgenic mice by a down-regulation of proapoptotic proteins and up-regulation of antiapoptotic proteins in the spinal cord tissue (Hwang and others 2009). VEGF delivered via an adenoassociated virus 9 in the SOD1 mice resulted in beneficial effects via the activation of the PI3-K/Akt survival pathway. In addition, VEGF reduced the production of the toxic M1 microglia and enhanced the differentiation of neuroprotective M2 microglial phenotype (Wang and others 2016).
The direct administration of VEGF in ALS and excitotoxic models also results in beneficial effects due to molecular mechanisms that require activation of VEGFR-2 (Storkebaum and others 2005). The intraperitoneal administration of VEGF reduced astrogliosis in the ventral horn of the spinal cord, preserved neuromuscular junctions, increased motor performance, delayed disease progression, and prolonged survival in ALS transgenic mice (Zheng and others 2004, 2007). Interestingly, in a rat model of spinal cord neurodegeneration, VEGF administration prevents hindlimb paralysis and motoneuron death induced by AMPA through VEGFR-2 and activation of PI3-K pathways, as well as inhibition of p38MAPK (Tovar-y-Romo and Tapia 2010).
Evidence for VEGF Deficiency and ALS in Humans
The human ALS literature provides contradictory evidence with respect to a role for VEGF. Spontaneous mutations of the HRE in the promoter region of the VEGF gene have not been detected in ALS individuals, as is the case for VEGFδ/δ mice. A meta-analysis of about 2000 subjects from Sweden, Belgium, and England detected that subjects homozygous for the _2578A/_1154A/_634G or _2578A/_1154G/_634G haplotypes in the VEGF gene promoter were more common in the population of patients with ALS than in healthy individuals. These “at-risk” haplotypes reduced the levels of VEGF in plasma and decreased VEGF gene transcription (Lambrechts and others 2003; Terry and others 2004). Other studies reported no association between the “at-risk” haplotypes and ALS in various populations from other countries (Del Bo and others 2008; Van Vught and others 2005; Zhang and others 2006). Subsequently, a meta-analysis was carried out to clarify the different results, including over 7000 subjects from eight European and three American populations. The results did not support the original conclusion that VEGF haplotypes increase the risk of ALS in humans, but the significant association of the low-VEGF –2578AA genotype with increased susceptibility to ALS in males supports the link between reduced VEGF concentrations and ALS (Lambrechts and others 2009).
The role of VEGF in human ALS due to promotor haplotypes is, therefore, variable and limited. It may be that VEGF deficiency causes ALS-like motoneuron degeneration but with the concern that all disorders involving motoneuron loss likely have similar symptomology. It is also possible that ALS may be caused by different factors in different individuals, with VEGF dysfunction only affecting a subset of patients with ALS. This could explain the variability in the results of the human genetic studies of ALS described above. Alternatively, other molecular aspects of VEGF could be altered in ALS, such as the responsiveness of its receptors, intracellular signaling pathways, or an autoimmune disorder. Reports on the levels of VEGF found in plasma, serum, and cerebrospinal fluid are also contradictory. Plasma VEGF levels were shown to be about 50% lower in individuals with ALS than in unaffected spouses (Lambrechts and others 2003). However, other studies have not found a difference in serum or plasma VEGF levels between patients with ALS and healthy individuals (Devos and others 2004). VEGF levels have been reported to be increased in serum from patients with ALS (Nygren and others 2002). Disease side effects such as platelet release of VEGF or hypoxia due to respiratory dysfunction might explain the variety of data reported. Therefore, plasma VEGF levels might initially be lower but subsequently increase due to hypoxia at later stages of the disease, complicating data interpretation (Lambrechts and others 2004).
VEGF levels in the cerebrospinal fluid of patients at an early stage of the disease have been reported to be low (Devos and others 2004). However, other studies report that VEGF is increased in the cerebrospinal fluid from patients with ALS (Gupta and others 2011; Ilzecka 2004). These contradictory results could be due to the small groups of patients and controls studied and to methodologic issues.
In an interesting study evaluating spinal cords of postmortem ALS and control individuals, immunohistochemistry procedures demonstrated a reduction in the staining for VEGF and VEGFR-2 in the neuropil of the anterior horn for the ALS cases. A reduced expression of VEGF and VEGFR-2 was confirmed by Western blotting and quantitative PCR. These results would support the hypothesis that reduced VEGF signaling might play a role in the pathogenesis of human ALS (Brockington and others 2006).
Mechanisms of Action of VEGF on ALS Animal Models
The presence of ALS-like symptoms due to the decreased VEGF levels in VEGFδ/δ mutant mice has led to two nonmutually exclusive hypotheses to explain the mechanism of action by which the deficiency of this factor produces motoneuron death: the vascular hypothesis and the neurotrophic insufficiency hypothesis. In the SOD1 transgenic mice, the mutant SOD1 protein competes with HuR for binding, destabilizing VEGF mRNA and thereby down-regulating VEGF synthesis. Thus, SOD1 mice also present low VEGF levels, and the motoneuron degeneration found in this model might be also explained, at least in part, by the same mechanisms proposed for VEGFδ/δ mice as schematized in Figure 4.
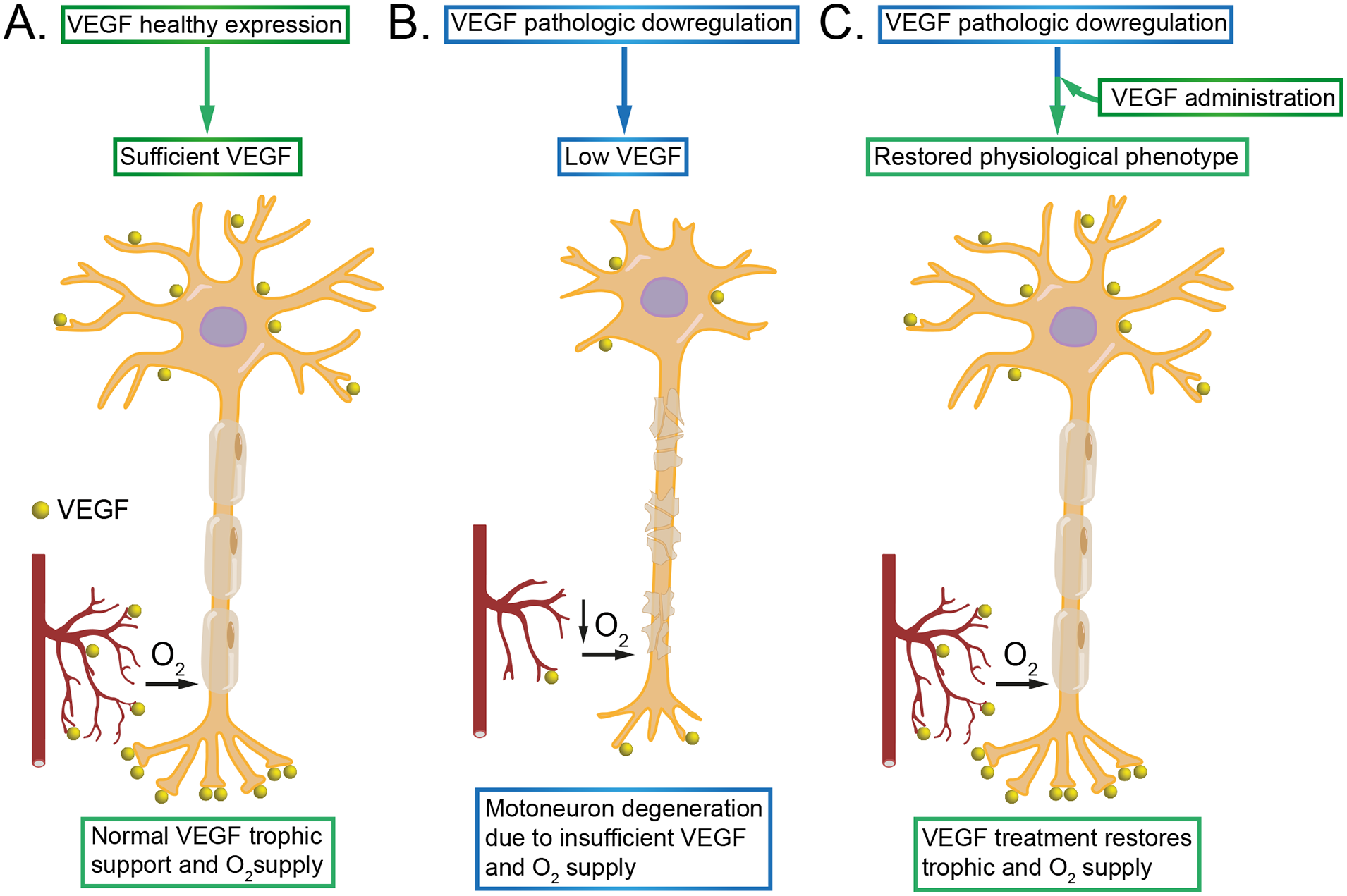
Proposed mechanisms of action of vascular endothelial growth factor (VEGF) in animal models of motoneuron degeneration. (A) Under healthy conditions, VEGF is adequately synthesized and acts on motoneurons by two likely mechanisms. First, due to its effects on the vasculature, motoneurons are provided with appropriate levels of oxygen and nutrients. Second, VEGF also acts as a neurotrophic factor for motoneurons. (B) In animal models of ALS, the synthesis of VEGF is reduced. The effects of low levels of VEGF on blood vessels produce a deficit in oxygen and nutrient supply to motoneurons. In addition, there is insufficient VEGF neurotrophic support. These two processes lead to motoneuron degeneration. (C) The potential therapeutic value of VEGF. It can be used as a strategy to promote motoneuron recovery. VEGF delivery (through different routes and methods) reestablishes normal levels of VEGF that would act on diseased motoneurons enhancing the supply of oxygen and nutrients via restored vascular function and by providing the required neurotrophic support, thus preventing motoneurons from degeneration.
The possibility of vascular abnormalities was investigated in VEGFδ/δ mice. Capillary densities and lumen sizes were normal in peripheral nerves and spinal cords, with no signs of leakiness, obstruction, or microangiopathy. However, reduced spinal cord perfusion seemed specific, as renal perfusion and muscle oxygenation were normal (Oosthuyse and others 2001). Chronic neuronal ischemia leads to oxidative stress, and motoneurons are especially vulnerable to oxidative damage, due to their large size, high metabolism, and distinct profile of glutamate receptors and calcium binding proteins (Shaw and Eggett 2000). Alternatively, the lack of a sufficient supply of oxygen and nutrients to motoneurons due to reduced vascular perfusion in mutant VEGFδ/δ mice might be enough to trigger the neurodegenerative process.
The second mechanism could be that motoneurons in VEGFδ/δ mice degenerate because there is insufficient VEGF-dependent neuroprotection, independent of angiogenic activity (Fig. 4). Oosthuyse and others (2001) studied the possible VEGF neuroprotective effect on cultured primary motoneurons of VEGF+/+ embryos, which lack vascular irrigation. The addition of VEGF to the culture increased survival of motoneurons by 20% in baseline conditions and by 50% after serum deprivation. VEGF also protected the mouse motoneuron-like NSC-34 cells against apoptosis induced by tumor necrosis factor α, hypoxia, oxidative stress (H2O2), or serum deprivation. The survival effect of VEGF required the activation of both VEGFR-2 and NRP-1, since the percentage of survival partially dropped by adding either anti–NRP-1 antibody or anti–VEGFR-2 antibody to the culture medium. A combination of anti–VEGFR-2 and anti–NRP-1 antibodies was required to completely neutralize the VEGF-dependent motoneuron survival activity (Oosthuyse and others 2001). Altogether, it is possible that motoneurons in VEGFδ/δ mice degenerate because of insufficient VEGF-dependent neuroprotection. Since VEGF is strongly and rapidly induced by hypoxia and other insults (e.g., acidosis), VEGF may represent a neuroprotective factor for motoneurons exposed to stress conditions. Therefore, motoneuronal VEGF-mediated neuroprotection may be relevant not only during hypoxia but also under other conditions that can result in a damage to motoneurons. For instance, excitotoxicity due to an excess of glutamate has been reported as one of the possible causes of ALS (Dhasmana and others 2022; Valko and Ciesla 2019), and VEGF administered in vivo protects motoneurons against this damage (Tovar-y-Romo and others 2007; Tovar-y-Romo and Tapia 2010).
Proof of a direct effect of VEGF in neuroprotection is the lack of angiogenesis caused by VEGF delivery (Azzouz and others 2004). Moreover, the intracerebrospinal administration of VEGF in SOD1 rats is also beneficial without inducing changes to blood vessel density or permeability, suggestive also of a direct neurotrophic effect of VEGF (Storkebaum and others 2005). In another study, VEGF was administered in a rat model of spinal cord injury, and the factor was demonstrated to rescue motoneurons from cell death with no changes in the vascular architecture of the spinal cord (Tovar-y-Romo and others 2007). Interestingly, a dose-dependent study of the effects of VEGF administration on ischemic rat brains demonstrated that doses that were too low to induce angiogenesis were still neuroprotective. In contrast, higher doses of VEGF, which induced angiogenesis, showed an absence of neuroprotection, perhaps due to the deleterious effects of brain edema and inflammation (Manoonkitiwongsa and others 2004).
In this respect, it is important to indicate that VEGF has contextual effects that can be both beneficial and deleterious, as has been particularly studied in stroke. Beneficial effects include an increase in collateral vessel formation, which can bypass occluded vessels and reduce infarct areas (reparative angiogenesis), and also neuroprotective mechanisms acting through neuronal VEGF receptors (Ma and others 2012; Zhang and others 2000). By contrast, high VEGF levels (particularly early after the infarct) may aggravate tissue damage through an increased blood-brain barrier leakage, leading to edema (Weis and Cheresh 2005). Edema increases intracranial pressure, which further increases ischemia by obstructing blood vessels (van Bruggen and others 1999; Zhang and others 2000). Moreover, increased vascular permeability also allows the entry of molecules and immune cells that do not normally enter into brain tissue due to the intact blood-brain barrier, causing neuroinflammation and homeostasis dysfunction (Geiseler and Morland 2018; Lange and others 2016). Such detrimental effects have, thus far, impeded the clinical use of VEGF to alleviate the sequels of ischemic stroke (Cárdenas-Rivera and others 2019). However, it should be stressed that experimental studies have reported beneficial effects in stroke when VEGF is administered after the acute phase (Geiseler and Morland 2018; Lange and others 2016; Zhang and others 2000). Thus, the dose, timing, and the route of administration are important factors to consider for evaluating the effectiveness of VEGF treatment in stroke (Geiseler and Morland 2018).
Neurotrophic Action of VEGF in Extraocular Motoneurons
Extraocular motoneurons are involved in the generation of eye movements and lie in three distinct brainstem nuclei: the oculomotor, the trochlear, and the abducens nuclei. The physiologic actions of VEGF have been studied in detail in the abducens motoneurons of the cat following lesion and exogenous VEGF administration, as well as after anti-VEGF antibody treatment in undamaged motoneurons. The abducens nucleus offers several advantages for the study of lesion-induced plasticity: the discharge pattern of abducens motoneurons is well characterized, and both their afferents and the signals they carry have been described in detail (Büttner-Ennever 2006; Davis-López de Carrizosa and others 2011; Delgado-García and others 1986; Escudero and Delgado-García 1988; Escudero and others 1992; Horn and Straka 2021). In addition, the firing activity of abducens motoneurons can be recorded using the alert chronic animal preparation, which allows a direct correlation of motoneuronal firing with eye movements under different experimental conditions (e.g., lesion, VEGF, or anti-VEGF antibody treatment).
Extraocular motoneurons are less vulnerable than other cranial or spinal motoneurons to degeneration in ALS (Haenggeli and Kato 2002; Reiner and others 1995). However, it is important to note that they also exhibit some signs of degeneration but at very late stages of the disease (Kimura and others 2014; Sharma and others 2011; Takahashi and others 1993; Tjust and others 2017).
Peripheral administration of VEGF to axotomized abducens motoneurons
Extraocular eye muscles contain VEGF (Calvo and others 2018; Silva-Hucha and others 2020), and thus the axotomy of abducens motoneurons leaves these cells deprived of their retrograde source of VEGF. Recordings of the discharge of these motoneurons in alert cats have revealed that (control) abducens motoneurons show a typical tonic-phasic firing pattern that correlates with eye position and eye velocity, respectively, during the different types of eye movements (Delgado-García and others 1986). When these motoneurons are axotomized, they exhibit an overall decrease in firing rate and a significant reduction in neuronal eye position and velocity sensitivities due, at least in part, to the loss of synaptic inputs (a process known as “synaptic stripping,” reviewed in Alvarez and others 2020), as first described by Delgado-García and others (1988) and later confirmed by other authors (Calvo and others 2018, 2020; Davis-López de Carrizosa and others 2009, 2010).
The chronic administration of VEGF to the proximal stump of the transected VIth (abducens) nerve prevents and recovers the synaptic loss and the firing alterations induced by axotomy. These findings were obtained after using two different protocols of administration. In the immediate administration protocol, VEGF first dose was applied immediately after lesion, preventing the appearance of axotomy-induced alterations. In the delayed administration protocol (VEGF first dose 20 days after lesion), VEGF produces recovery of the synaptic and physiologic changes already present in severed motoneurons (Calvo and others 2018). It was also demonstrated that the tyrosine kinase receptors of VEGF (i.e., VEGFR-1 and VEGFR-2) mediate different roles in the maintenance of synaptic afferents to abducens motoneurons. It should be mentioned that, in contrast to VEGF, the administration to axotomized abducens motoneurons of the neurotrophins BDNF, NT-3, or NGF induces only partial recovery of synaptic inputs and discharge activity (Davis-López and others 2009, 2010).
Interestingly, the administration of VEGF-B, using the immediate administration protocol, also prevents the modifications induced by injury in abducens motoneurons (Calvo and others 2018). Previous results have also reported a neuroprotective role for VEGF-B in spinal motoneurons. Thus, VEGF-B protects cultured primary motoneurons against degeneration. Moreover, mice lacking VEGF-B also develop a more severe form of motoneuron degeneration when intercrossed with mutant SOD1 mice. When delivered intracerebroventricularly, VEGF-B prolongs the survival of mutant SOD1 rats (Poesen and others 2008). Altogether, these results may have an important therapeutic value, because VEGF-B is virtually nonangiogenic and, therefore, its administration in the case of motoneuronal disorders could produce beneficial effects without the side effects of a high dose of VEGF, such as tissue edema and inflammation caused by excessive angiogenesis and vascular permeability (Calvo and others 2018; Manoonkitiwongsa and others 2004; Poesen and others 2008).
Several types of brainstem and spinal motoneurons down-regulate the expression of choline acetyltransferase when axotomized (reviewed by Navarro and others 2007; see also Morcuende and others 2005, 2013). Thus, peripheral administration of VEGF to axotomized extraocular motoneurons in adult rats also prevents the loss of their cholinergic phenotype (Acosta and others 2018). The immunostaining, enzymatic activity, and mRNA levels of choline acetyltransferase are also reduced in spinal motoneurons of patients with ALS (Nagata and others 1982; Oda and others 1995; Virgo and others 1992). All these findings suggest that VEGF administered to diseased motoneurons not only rescues them from cell death (as described above) but also maintains their neurotransmissive phenotype, synaptic inputs, and discharge pattern (Acosta and others 2018; Calvo and others 2018).
Intraventricular delivery of a single dose of VEGF to axotomized abducens motoneurons
The administration of VEGF into the IVth ventricle, located dorsal to the abducens nucleus, just after the section of the VIth nerve, also prevents the axotomy-induced alterations in the discharge pattern and synaptic inputs of damaged abducens motoneurons, as demonstrated by single-unit extracellular recordings and immunocytochemistry (Calvo and others 2020).
In addition, the study of the vasculature after VEGF intraventricular delivery revealed absence of angiogenesis in the abducens nucleus, as well as in other brainstem nuclei (Calvo and others 2020). Absence of angiogenesis and vascular leakage has also been shown after the peripheral administration of VEGF in the three extraoculomotor nuclei after axotomizing all extraocular motoneurons in adult rats (Acosta and others 2018). These findings indicate that the beneficial effects obtained with VEGF likely result from a direct action of the factor on the motoneurons themselves, rather than by an indirect action on the vascular system (Acosta and others 2018; Calvo and others 2020). A recent study using an anti-VEGF antibody, to reduce the retrograde delivery and transport of this factor, has shown that control, treated extraocular motoneurons transformed into the lesioned state, both in terms of firing and synaptic coverage (Calvo and others 2022).
A schematic drawing is illustrated in Figure 5 to compare the changes in synaptic inputs and firing of abducens motoneurons in the three conditions: control (Fig. 5A), axotomy (Fig. 5B), and anti-VEGF antibody treatment (Fig. 5C). The data obtained in extraocular motoneurons demonstrating the neurotrophic effects of VEGF (Acosta and others 2018; Calvo and others 2018, 2020, 2022) are consistent with all the evidence summarized in the present review supporting the neuroprotective effects of VEGF that have previously been studied mainly in spinal motoneurons from animal models of motoneuron disease.
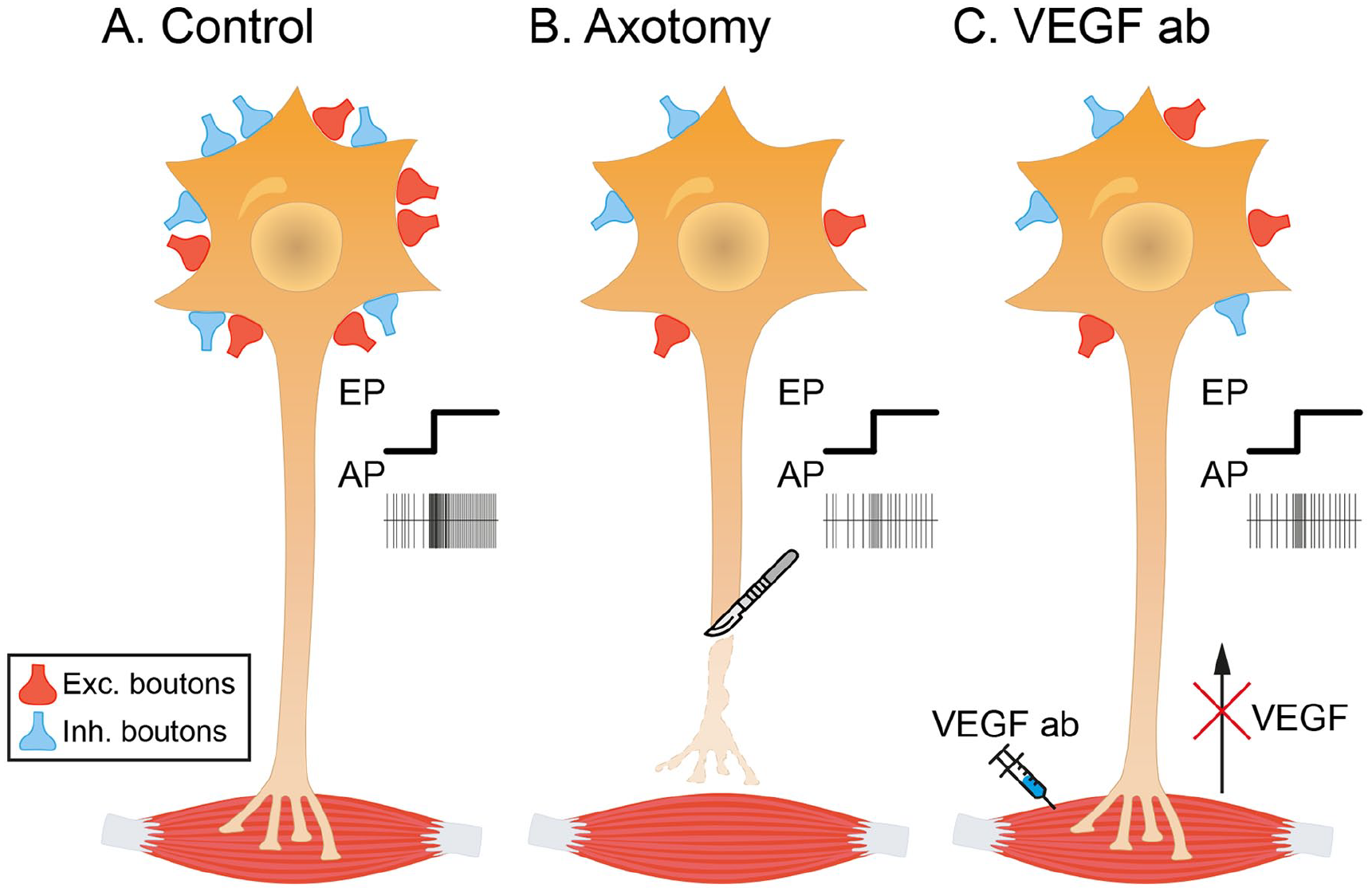
Schematic representation summarizing the effects of the administration of anti–vascular endothelial growth factor (VEGF) antibody to abducens motoneurons on their discharge activity and synaptic inputs. (A) Control motoneurons receive abundant synaptic boutons contacting their cell body, of both excitatory (in red) and inhibitory (in blue) nature. They exhibit a typical tonic-phasic discharge pattern of action potentials (APs) in correlation with eye movements (EPs, eye position). (B) After axotomy, the motoneuron experiences a significant loss of afferent synaptic boutons (excitatory and inhibitory), a phenomenon known as synaptic stripping. In parallel, the discharge pattern of axotomized motoneurons is markedly reduced. (C) Uninjured motoneurons treated with the neutralizing antibody against VEGF (VEGF ab) applied in the muscle lack their retrograde source of VEGF. Antibody-treated motoneurons also show synaptic stripping and a reduced firing pattern in correlation with eye movements, resembling the axotomy situation.
Conclusions
VEGF was discovered due to its angiogenic activity, but strikingly, it appears first in evolution as a neurotrophic factor, necessary for the appropriate development of the nervous system in invertebrates lacking a vascular system or having a rudimentary vasculature. Neuroscience interest in VEGF has grown remarkably in recent years due to mounting evidence showing that VEGF is neuroprotective. Mutant mice characterized by low levels of VEGF (VEGFδ/δ) have severe adult-onset muscle weakness and motoneuron degeneration, symptoms that resemble ALS. Experiments using crossbreeding between transgenic mice have reinforced the role of VEGF as an essential factor for motoneuron survival. Thus, when SOD1 mice (classical animal model of ALS) are crossed with mice overexpressing VEGF (VEGF+/+), the double transgenic animals show reduced motoneuron degeneration and a longer life expectancy. The opposite occurs when VEGFδ/δ and SOD1 mice are crossed, as the double mutants show earlier and more severe motoneuron degeneration and muscle weakness.
Recent results in extraocular motoneurons also point to a role of VEGF in neuroprotection since its administration through the proximal stump of the sectioned nerve or intraventricularly prevents motoneuron degeneration, produces structural and functional recovery of changes induced by the lesion, and can return axotomized motoneurons to their normal operating mode. A striking result has recently been shown in this model consisting of the administration of VEGF neutralizing antibody to intact, undamaged extraocular motoneurons. These results show that antibody-treated motoneurons resemble injured motoneurons, in that they discharge abnormally at low frequencies and also experience synaptic loss.
All the findings summarized in the present review indicate that VEGF is an essential neurotrophic factor for the survival and normal physiologic function of motoneurons. They further suggest that this factor might have a high potential therapeutic value in the treatment of motoneuron disorders. Future research could be directed at exploring possible routes of administration, applied dose, timing of application, and the use of vectors to deliver the factor chronically in nonhuman primates with motoneuron dysfunction. Another promising factor for the treatment of motoneuron disorders is VEGF-B, which is not angiogenic, since it is also neuroprotective with the advantage of lacking the side effects of VEGF. Nevertheless, further investigation on VEGF-B would be necessary to consider this factor as a useful clinical tool.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This publication is part of the I+D+i project P20_00529 Consejería de Transformación Económica Industria y Conocimiento, Junta de Andalucía-FEDER. Research materials were also supported by project PGC2018-094654-B-100 and PID2021-124300NB-I00 funded by MCIN/AEI/FEDER “A way of making Europe.” P.M.C. was a scholar of Ministerio de Educación y Ciencia (BES-2016-077912) in Spain and is now a “Margarita Salas” postdoctoral fellow. R.G.H. is a postdoctoral fellow from PAIDI-2019, “Talento Doctores” Junta de Andalucía in Spain. Confocal images were performed in the Central Research Services of Universidad de Sevilla.