
Abstract
Our previous studies demonstrated coordinate expression of platelet-derived growth factor (PDGF) -B chain and β-receptor in neurons at risk in the rat brain with focal ischemia. To clarify a role of the -B chain in the brain further, we examined whether PDGF-A or -B chain protects CA1 pyramidal neurons from delayed neuronal death after forebrain ischemia in rats. Pretreatment with PDGF-BB, but not -AA, at 120 ng/d for 2 days until forebrain ischemia was performed markedly ameliorated delayed neuronal death in CA1 pyramidal neurons on day 7 after ischemia. This neuroprotective effect of PDGF-BB was dose-dependent, and pretreatment with PDGF-BB at 240 ng/d showed almost complete inhibition of delayed neuronal death. In contrast, posttreatment with PDGF-BB at 120 ng/d starting 20 minutes after ischemia demonstrated no significant neuroprotective effect. The current study established marked neuroprotective actions of PDGF-BB in ischemic neuronal damage.
Keywords
Platelet-derived growth factor (PDGF) was originally described as a potent mitogen and chemoattractant for fibroblasts, glial cells, and smooth muscle cells (Ross et al., 1986; Heldin and Westermark, 1990; Raines et al., 1990). Platelet-derived growth factor consists of two related polypeptide chains (A and B), which are encoded by different genes located on different chromosomes. They are assembled naturally as a heterodimer (PDGF-AB) and two homodimers (PDGF-AA and PDGF-BB) (Ross et al., 1986; Heldin and Westermark, 1990; Raines et al., 1990). The biologic activities of PDGF are mediated through binding to two cell-surface receptors designated a and β, both of which have intrinsic tyrosine kinase activity. The α subunit can bind to either the -A or -B chain, whereas the β subunit can bind only to the -B chain (Hart et al., 1988; Heldin et al., 1988; Matsui et al., 1989; Seifert et al., 1989).
In the central nervous system, the PDGF-A chain was shown to play an important role in the proliferation and programed differentiation of oligodendrocyte-type 2 astrocyte progenitor cells of the glial lineage (Noble et al., 1988; Raff et al., 1988). However, it was shown recently that both PDGF-A and -B chains are present in neuronal cells in the developing and mature central nervous system, suggesting a crucial role of PDGF as a neuronal regulatory agent (Sasahara et al., 1991; Yeh et al., 1991; Sasahara et al., 1992; Hutchins and Jefferson, 1992). In addition, both PDGF α- and β- receptor mRNAs were shown to be widely expressed in the brain (Sasahara et al., 1991). The α-receptor mRNA in the brain was found to be expressed largely in glial cells during development in the mouse and rat (Pringle et al., 1992; Yeh et al., 1993). The β-receptor is expressed at high levels in the mouse forebrain during embryonic and early postnatal development (Hutchins and Jefferson, 1992). A previous study showed that PDGF-B chain has trophic activity on cultured neurons through binding to the β-receptor (Smits et al., 1991). Taken together, these observations suggest that PDGFs may affect not only glial cells but also neurons through binding to their high affinity receptors on these cells mainly during the embryonic and early postnatal development of the brain. However, the roles of PDGFs, especially in the mature brain, remain unknown.
Recently, we have shown that ischemia enhances expression of -B chain mRNA and increases -B chain immunoreactivity in neurons and brain macrophages in the focal ischemia model in the rat brain (Iihara et al., 1993, 1994). In contrast, PDGF-A chain mRNA expression remains unchanged in the ischemic brain, suggesting an important role of the -B chain in the ischemic brain (Iihara et al., 1994). Furthermore, the expression of PDGF β-receptor, which is specific to the -B chain, was found to increase in the ischemic brain, supporting the hypothesis that PDGF-B chain may play a crucial role in the cellular cascade of the healing process in the ischemic brain, including survival of neurons at risk, glial response, accumulation of brain macrophages, and neovascularization (Iihara et al., 1996a). The present study was designed to determine whether PDGFs can rescue the delayed neuronal death after transient forebrain ischemia in rats. Preliminary results of this study were published previously in abstract form (Iihara et al., 1996b).
MATERIALS AND METHODS
Induction of forebrain ischemia
Male Wistar rats, weighing 180 to 220 g, were used for the experiments. The animals were starved during the night preceding the induction of forebrain ischemia but were allowed water ad libitum. Transient forebrain ischemia was induced according to the method reported by Smith et al. (1984) with some modifications (Kaku et al., 1993). Briefly, rats were anesthetized with 3.5% halothane, intubated, and connected to a Starling-type respirator. Anesthesia was maintained with 0.7% halothane and 30% O2 in N2O. The femoral artery and vein were exposed to insert polyethylene catheters for monitoring arterial blood pressure, arterial blood gases, and blood glucose level. After isolation of the bilateral common carotid arteries, halothane was discontinued and the rats were allowed to remain in a steady state for a period of 20 minutes, with the rectal temperature at 37°C, Pa
Reproducibility of delayed neuronal death in the pyramidal neurons in CA1 subfield was examined in rats (n = 7) subjected to transient forebrain ischemia alone without implantation of an osmotic minipump.
Experiment 1. Effects of PDGF pretreatment on survival of pyramidal neurons in CA1 subfield after forebrain ischemia
Rats were divided into three experimental groups (n = 7 each). In each group, an osmotic minipump (Alza model 2002, 200-μL fill volume, 0.5-μL/h pump rate, Alza Corp., Palo Alto, CA) contained (I) bovine serum albumin (BSA, Fraction V, Sigma Chemical Co., St. Louis, MO, U.S.A.) as a control, (II) recombinant human PDGF-AA (Oncogene Science), or (III) recombinant human PDGF-BB (Oncogene Science). In each group, the minipump contained 2 μg of the respective agent, dissolved in 200 μl of artificial CSF that had been prepared in a sterile, pyrogen-free water.
The rats were anesthetized with pentobarbital sodium (50 mg/kg intraperitoneally) and placed in a stereotactic head holder. The coordinates of stereotactic puncture into the right lateral ventricle were set as 0.8 mm posterior to the bregma, 1.5 mm right of the midline, and 4.3 mm beneath the surface of the skull. The inserted 29-gauge stainless steel intraventricular cannula was fixed to the skull with bone cement and connected to a polyethylene tube. The osmotic minipump was implanted subcutaneously in the neck and connected to the tube. At the concentration and pump rate used, BSA, PDGF-AA, or PDGF-BB were delivered at a dose of 120 ng/d into the right lateral ventricles of normothermic rats until the transient forebrain ischemia procedure was performed.
Two days after beginning the infusion, rats were subjected to transient forebrain ischemia as described (Kaku et al., 1993) after removal of the intraventricular cannula.
On day 7 after ischemia, the rats were deeply anesthetized with pentobarbital, and perfused transcardially with 0.05 mol/L phosphate-buffered saline at pH 7.4 and then with 2% paraformaldehyde in the same buffer. The perfused brain was dissected out, cut into coronal slices 5-mm thick at the level of the dorsal hippocampus, and incubated in the same fixative overnight. Frozen coronal sections 20-μm thick were then prepared and stained with hematoxylin and eosin. Under the light microscope at a magnification of ×400, the numbers of surviving pyramidal neurons in the center of the right CA1 subfield per 1 mm were counted. All cell counting was done blind with respect to experimental groups.
The numbers of the surviving pyramidal neurons and physiologic parameters in the PDGF-AA or -BB-treated groups were compared with those of rats subjected to continuous infusion of BSA (Control) with the ischemic procedure (n = 7 each).
Experiment 2. Dose-response analysis of the neuroprotective effects of pretreatment with PDGF-AA or BB on delayed neuronal death after forebrain ischemia
Platelet-derived growth factor-AA or -BB at a dose of 60 or 240 ng/d were infused intracerebroventricularly for 2 days until the onset of the transient forebrain ischemia procedure in normothermic rats as described in experiment 1 (n = 7 each). Direct visual counting of the surviving CA1 pyramidal neurons was performed under a microscope. All cell counting was done blind with respect to experimental groups. Control animals received continuous intracerebroventricular infusion of BSA.
Experiment 3. Effects of posttreatment with PDGF-BB on survival of pyramidal neurons in CA1 subfield after forebrain ischemia
Male Wistar rats (n = 7) weighing 180 to 220 g were subjected to transient forebrain ischemia after overnight fasting as described above. Twenty minutes after cessation of forebrain ischemia, rats were placed in a stereotactic head holder and subjected to continuous intracerebroventricular infusion of PDGF-BB as described above (120 ng/mL) until day 7 after ischemia. On day 7, rats were sacrificed under deep anesthesia, and the numbers of pyramidal neurons per 1 mm in the center of CA1 subfield were counted. All cell counting was done blind with respect to experimental groups. Control rats (n = 7) were subjected to transient forebrain ischemia with continuous intracerebroventricular infusion of BSA (120 ng/mL) otherwise in the same procedure.
Immunostaining for glial fibrillary acidic protein
To examine the involvement of glial cells in the neuroprotective actions of PDGF-BB, immunostaining for glial fibrillary acidic protein (GFAP) was done on the coronal section at the level of insertion of the catheters in groups that had been subjected to continuous infusion of PDGF-AA or -BB for 2 days (120 ng/mL; n = 3 each) without induction of transient forebrain ischemia. Glial fibrillary acidic protein immunostaining was done as described in our previous paper (Iihara et al., 1996a). The numbers of GFAP-positive cells around the site of insertion of the catheters into the ventricle was counted in the high-power field under microscope.
Statistical analysis
Data are expressed as means ± SD. The significance of the results was tested with one-way analysis of variance followed by Fisher's protected least significance difference for multiple comparisons between groups. Differences were considered statistically significant when the P value was < 0.05.
RESULTS
Physiologic parameters, including blood glucose levels, arterial pH, Pa
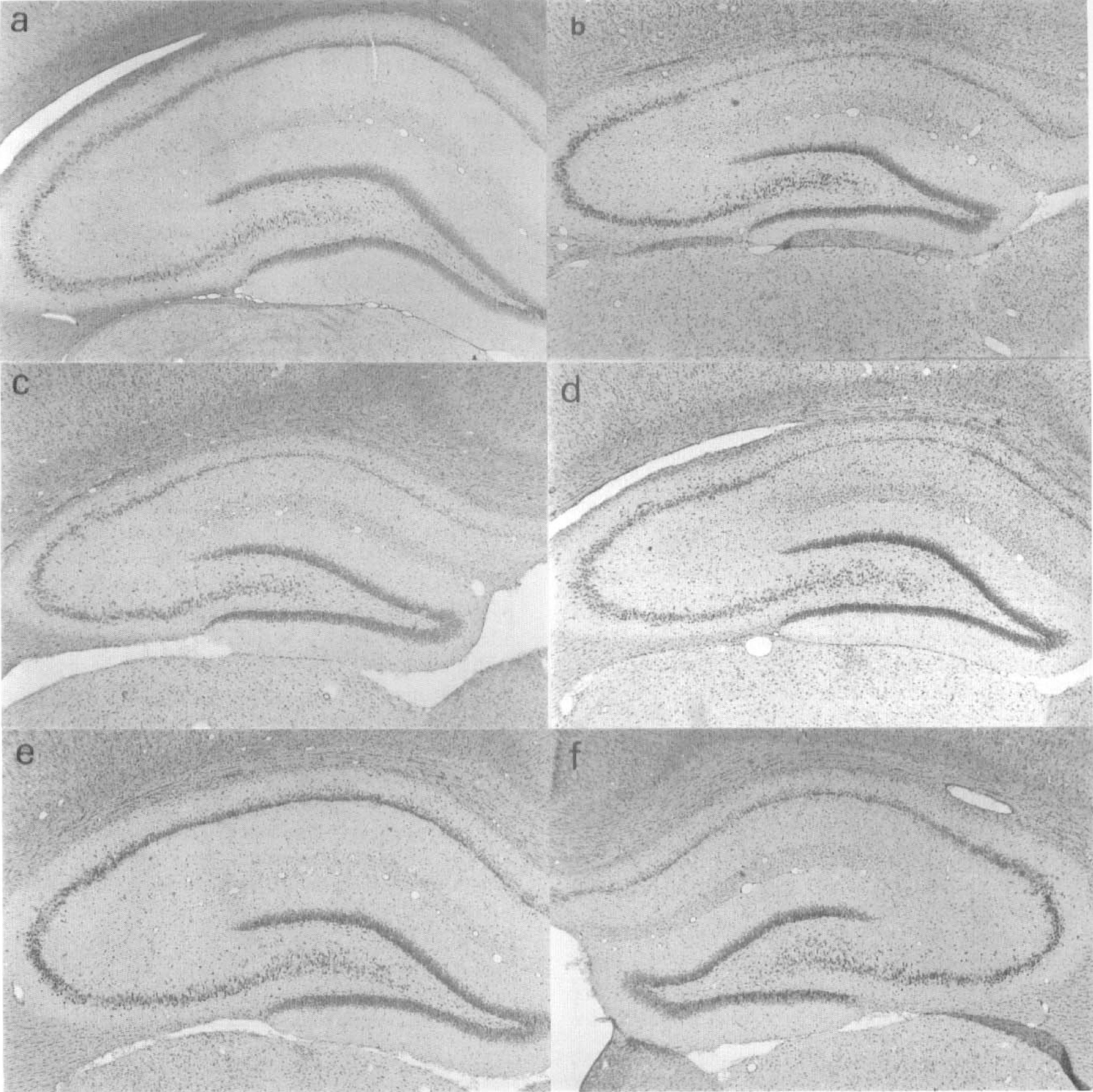
Pretreatment with platelet-derived growth factor (PDGF)-BB, but not -AA, ameliorated delayed neuronal death in CA1 pyramidal neurons in the hippocampus after forebrain ischemia in rats. (
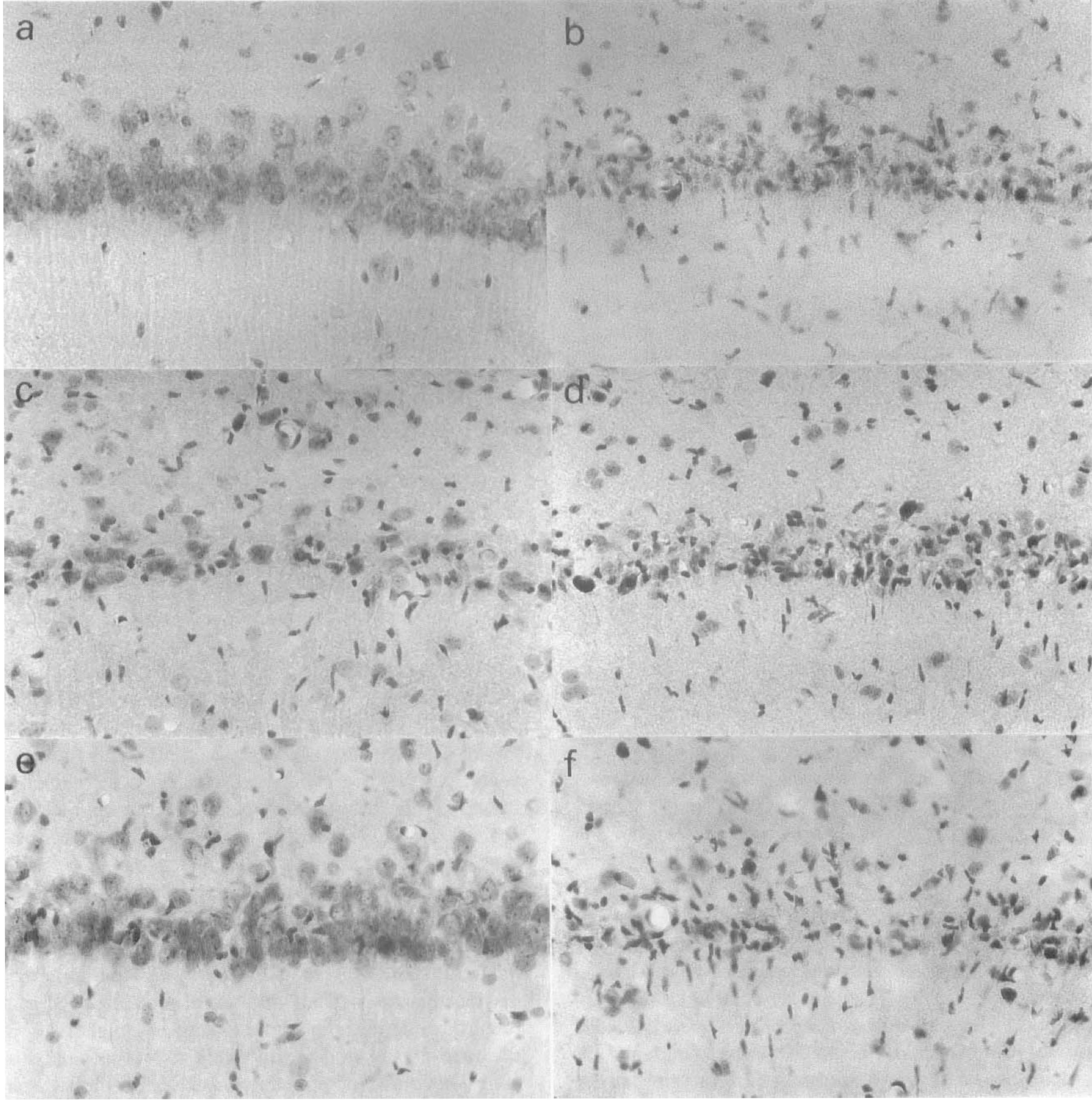
Higher magnification photomicrographs (400×) of pyramidal neurons in the CA1 subfield. Note that many pyramidal neurons in the CA1 subfield were rescued by pretreatment with PDGF-BB(
Physiologic variables
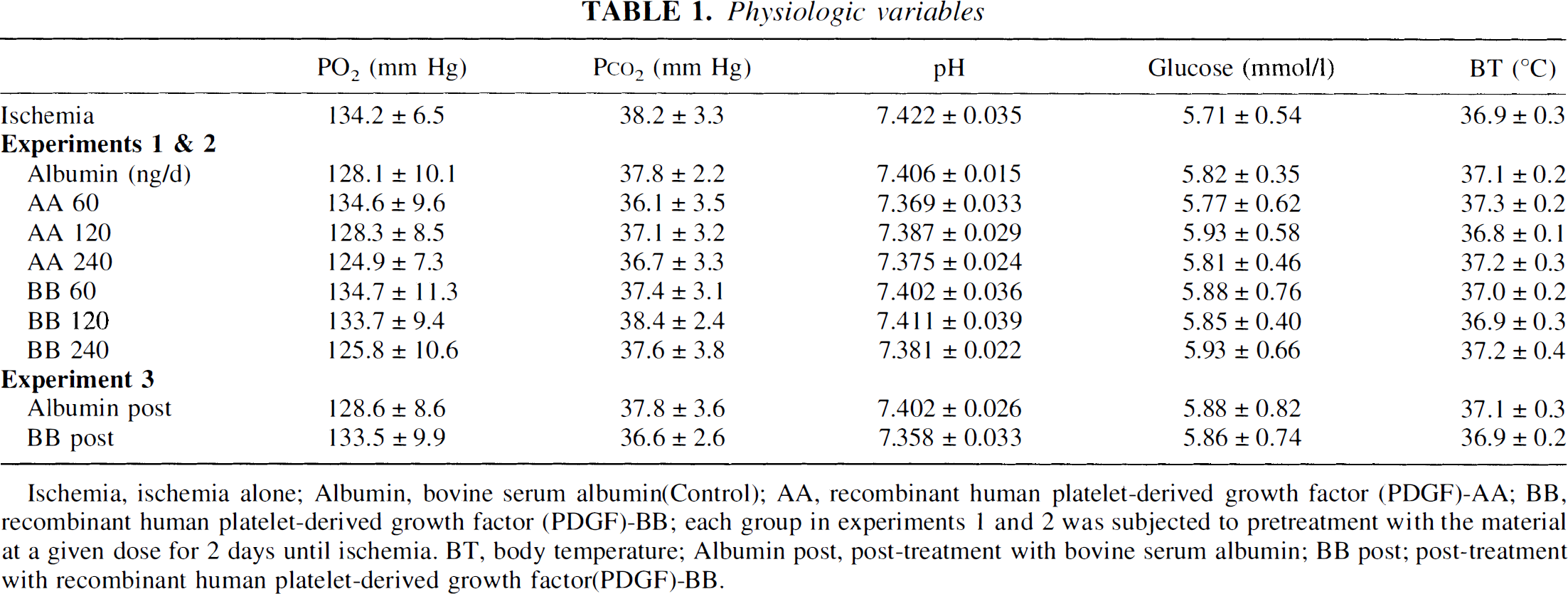
Ischemia, ischemia alone; Albumin, bovine serum albumin(Control); AA, recombinant human platelet-derived growth factor (PDGF)-AA; BB, recombinant human platelet-derived growth factor (PDGF)-BB; each group in experiments 1 and 2 was subjected to pretreatment with the material at a given dose for 2 days until ischemia. BT, body temperature; Albumin post, post-treatment with bovine serum albumin; BB post; post-treatment with recombinant human platelet-derived growth factor(PDGF)-BB.
Pretreatment with PDGF-AA at 120 ng/d showed no significant neuroprotective effects against delayed neuronal death in the CA1 subfield as compared with the BSA group. The numbers of pyramidal neurons in the hippocampal CA1 subfields on day 7 were 46.1 ± 7.3/mm and 53 ± 5.9/mm, respectively (Figs. 1C, 1D, 2C, and 2D). In contrast, PDGF-BB at a dose of 120 ng/d markedly ameliorated delayed neuronal death in the CA1 pyramidal neurons in the right hippocampus on day 7 after ischemia (252.8 ± 78.7/mm; n = 7, P < 0.001 vs. BSA-treated animals) (Figs. 1E and 2E). This reduction in neuronal death was confined to the hippocampus ipsilateral to the ventricular infusion, and no significant increase was observed in the numbers of surviving CA1 pyramidal neurons in the left hippocampus (Figs. 1F and 2F).
Three different concentrations of PDGF-AA or PDGF-BB were tested and significant increases in survival of CA1 pyramidal neurons were seen by pretreatment with PDGF-BB at doses of 120 and 240 ng/d (252.8 ± 78.7/mm; n = 7, P < 0.001; 354.5 ± 36.0/mm; n = 7, P < 0.001; respectively vs. BSA-treated animals) (Figs. 3A and 4). The most pronounced reduction of neuronal death was observed at a dose of 240 ng/d, and administration of PDGF-BB at this dose showed almost complete inhibition of delayed neuronal death in the CA1 pyramidal neurons in the right hippocampus on day 7 after ischemia (354.5 ± 36.0/mm, n = 7) (Fig. 3A). Even at this dose, the neuroprotective effect was seen only in the right hippocampus (data not shown). On the other hand, no significant reduction in neuronal death was found by pretreatment with PDGF-AA at doses up to 240 ng/d (Fig. 4).
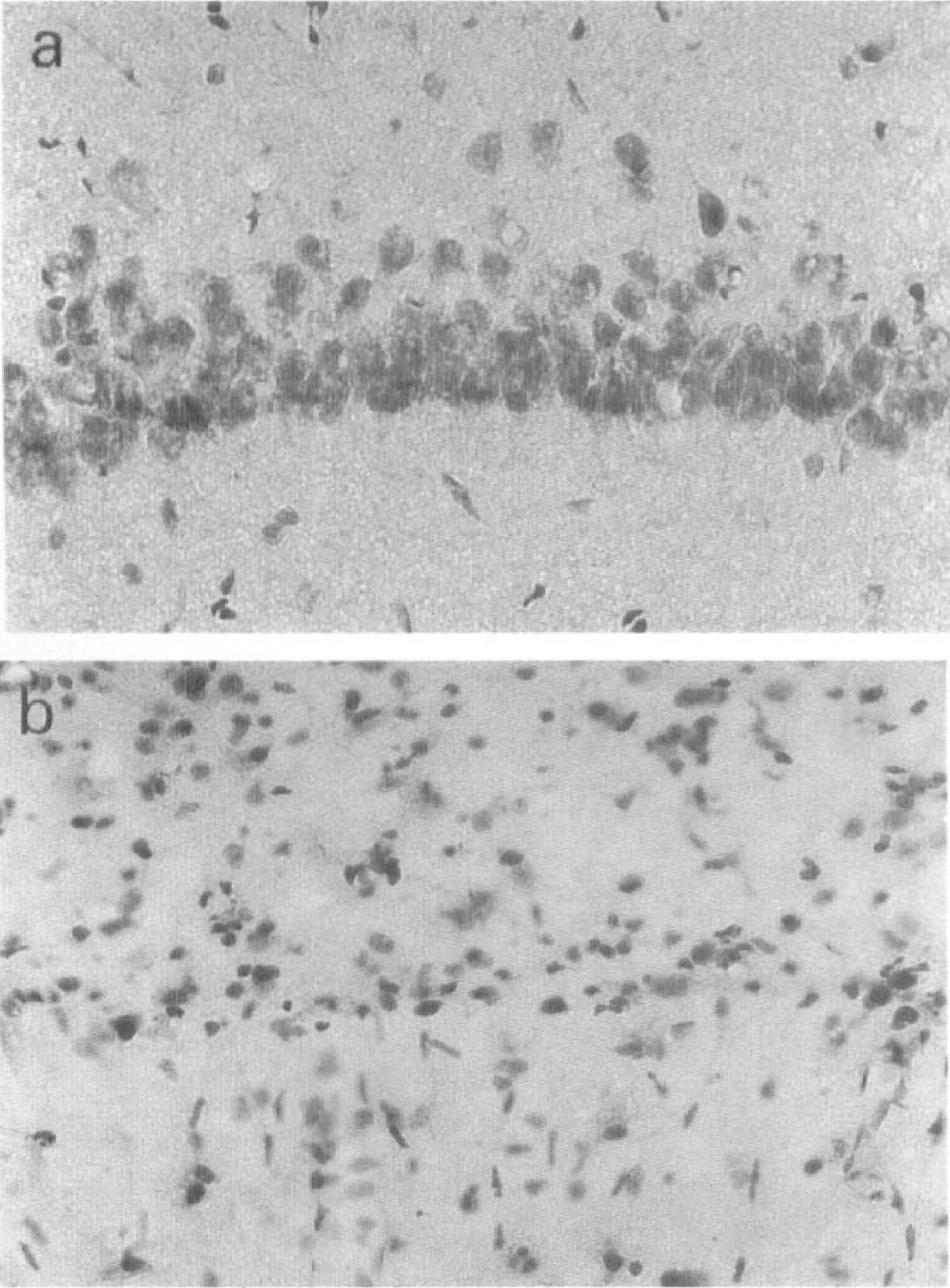
Higher magnification photomicrographs of CA1 pyramidal neurons (×400) after forebrain ischemia in rats subjected to (a) pretreatment with PDGF-BB (240ng/d) and (b) post-treatment with PDGF-BB (240ng/d). Pretreatment with PDGF-BB at dose of 240 ng/d for 2 days almost completely prevented delayed neuronal death, but post-treatment with PDGF-BB at the same rate of infusion for 7 days failed to rescue the neurons.
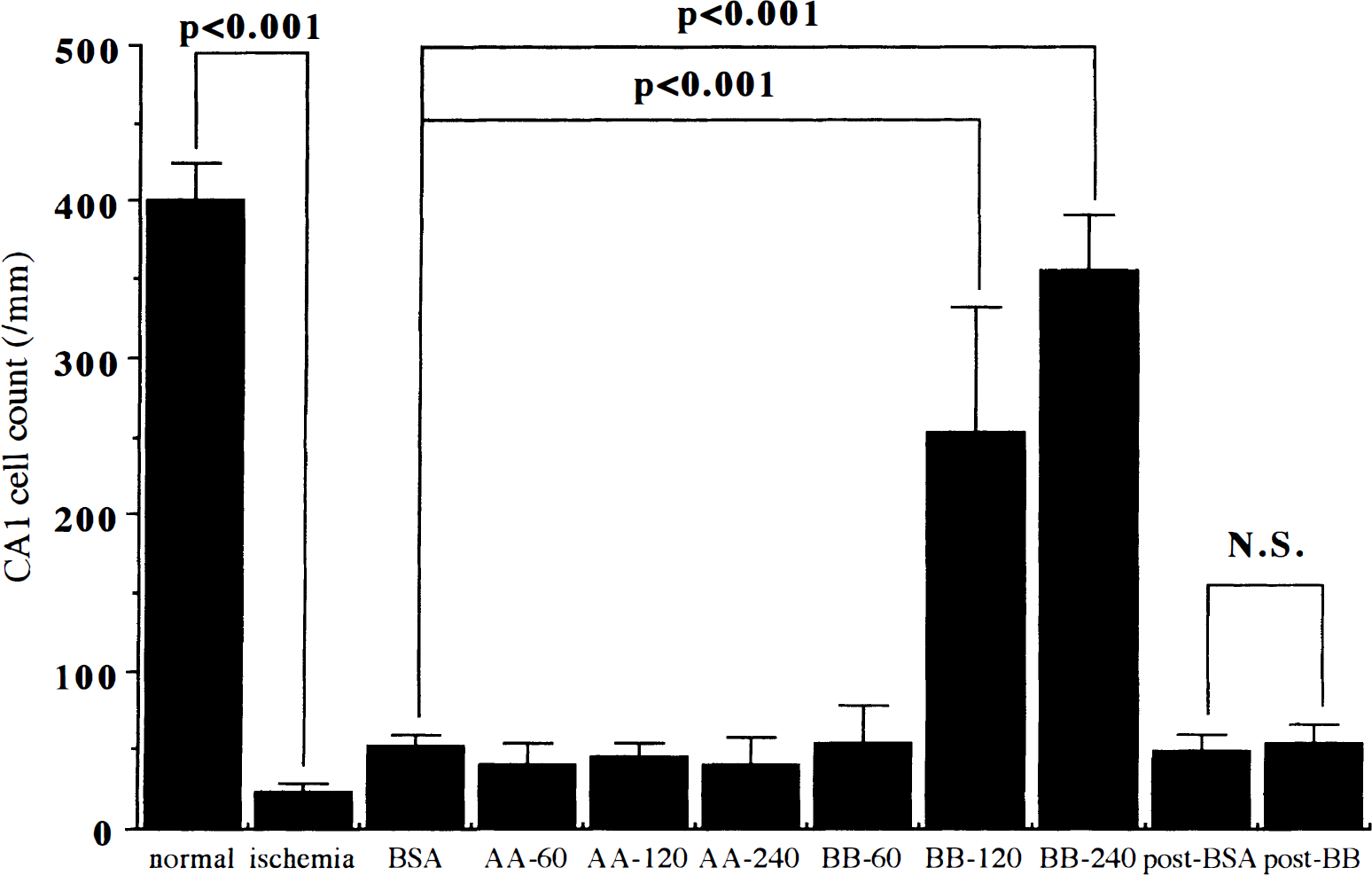
Bar graph showing dose-dependency of neuroprotective actions of pretreatment with PDGF-BB against delayed neuronal death in the pyramidal neurons in the CA1 subfield. Significant and dose-dependent neuroprotective actions were observed in the groups subjected to pretreatment with PDGF-BB at doses of 120 and 240 ng/d for 2 days until ischemia (one-way analysis of variance followed by Fisher's protected least significance difference [PLSD]). In contrast, neither pretreatment with PDGF-AA at doses up to 240 ng/d or posttreatment with PDGF-BB (120 ng/d) showed significant neuroprotective effects. Data are expressed as mean ± SD (n = 7 each). BSA, bovine serum albumin; N.S., not significant.
The effects of PDGF-BB at 120 ng/d on the delayed neuronal death were also tested in rats subjected to post-treatment from 20 minutes until day 7 after ischemia. No significant increase in the survival of CA1 pyramidal neurons was observed on day 7 after ischemia at this dose of PDGF-BB (54.0 ± 24.0/mm, n = 7) as compared with BSA (53.0 ± 5.9/mm, n = 7) (Figs. 3B and 4).
The immunostaining for GFAP in the rats that had been infused continuously for 2 days (120 ng/d) showed no significant difference of GFAP-positive cells in the vicinity of the ventricular wall that had been penetrated by the catheters (Fig. 5)

Immunostaining for glial fibrillary acidic protein (GFAP) around the site of insertion of the catheters into the lateral ventricle showed no significant difference in the number of GFAP-positive cells between PDGF-AA and -BB-treated groups. Original magnification ×200.
DISCUSSION
The present study demonstrated that pretreatment with PDGF-BB, but not PDGF-AA, showed a marked neuroprotective effect against delayed neuronal death of pyramidal neurons in the CA1 subfield after forebrain ischemia in rats. This neuroprotective effect was dose-dependent, and almost complete inhibition of delayed neuronal death was seen at a dose of 240 ng/d for 2 days before ischemia. However, no such reduction in neuronal death was observed by posttreatment with PDGF-BB.
Recently, several neurotrophic or growth factors, such as nerve growth factor (NGF), brain-derived neurotrophic factor, acidic and basic fibroblast growth factors, and ciliary neurotrophic factors, have been reported to reduce delayed neuronal death after transient forebrain ischemia in rodents (Shigeno et al., 1991; Yamamoto et al., 1992; Sasaki et al., 1992; Nakata et al., 1993; Tsukahara et al., 1994; Beck et al., 1994; Wen et al., 1995). In contrast, other groups reported no neuroprotective effects of these factors (Hara et al., 1991; Beck et al., 1992). These inconsistent results may be explained by differences in dose, rate, and timing of infusion used in these studies (Yamamoto et al., 1992).
The dose and rate of infusion chosen in the present study were similar to those in some previous studies using acidic and basic fibroblast growth factors, and ciliary neurotrophic factor (240 ng/d, 240 ng/d, and 50–500 ng/d, respectively, continuous intraventricular infusion) (Sasaki et al., 1992; Nakata et al., 1993; Wen et al., 1995), but much less than in another study using NGF (10 μg bolus, intraventricular injection) (Shigeno et al., 1991). In the present study, significant neuroprotection with PDGF-BB was found to be limited to the hippocampus on the side ipsilateral to the ventricular infusion. The reason for this lack of neuroprotection in the contralateral hippocampus remains unknown, but may be explained by the lower concentration of the factor in the contralateral ventricle or by the more limited diffusion into the hippocampus in the hemisphere not injured by placement of the catheter. At the present time, it is unclear how far PDGF is capable of diffusing through traumatized or nontraumatized brains. In case of intraventricular infusion of NGF, some studies indicated that NGF can diffuse through the brain as little as 1 mm (Lapchak et al., 1993), whereas other studies reported specific uptake and transport of NGF to basal forebrain cholinergic system in nonhuman primate (Emmet et al., 1994). It was also reported that there was a good correlation between the infused NGF labeling pattern and the presence of trk A NGF receptors (Lapchak et al., 1993). Platelet-derived growth factor, infused into the ventricle, seems to reach the contralateral ventricle as rapidly as is reported for NGF (Lapchak et al., 1993). On the other hand, our previous studies demonstrated enhanced immunoreactivity for PDGF β-receptor in the neurons at risk and reactive glial cells in the vicinity of infarct and stab wound in the rat brain (Iihara et al., 1996a: Iihara et al., unpublished observation). Taken together, these studies suggest that the increased expression and altered localization of PDGF receptor after insertion of the catheter might have changed the uptake mechanism and accumulation of infused PDGF. To conclude definitely on this issue and determine the ideal dose and rate of infusion of PDGF-BB, more studies are necessary on their uptake mechanisms, time course of intraventricular and intrahippocampal concentration of PDGF, and the expression of PDGF receptors after the ventricular infusion.
Our results demonstrated no neuroprotective effects by posttreatment with PDGF-BB beginning 20 minutes after ischemia at the concentration employed (120 ng/d). In contrast, some previous studies reported a neuroprotective effect on delayed neuronal death after forebrain ischemia by posttreatment with a single bolus injection of a much higher dose of NGF (10 μg) at 15 minutes after ischemia (Shigeno et al., 1991) or with delayed continuous infusion of acidic fibroblast growth factor, starting earlier (5 minutes) after ischemia (Sasaki et al., 1992) as compared with the current study. Ginsberg (1995) suggested that there may be a continuum of therapeutic benefit of a given agent such that the extent of benefit diminishes gradually as the time to initiation is prolonged and the intensity of treatment decreases. Because these neurotrophic or growth factors share some neuroprotective actions on the cascade of events elicited after ischemic insult as discussed below, delayed and insufficient increases in the intraventricular concentration of PDGF-BB at the dose used in the present study may account for the lack of effects against delayed neuronal death. An alternative explanation for the lack of effect of posttreatment with PDGF-BB is that the sustained inhibition of protein synthesis after global ischemia (Thilmann et al., 1986; Widmann et al., 1991) may preclude de novo translation of essential, neuroprotective proteins induced by PDGF, although at least some proteins, such as several immediate early gene (Jorgensen et al., 1991; Neumann-Haefelin et al., 1994) and heat shock proteins (Chopp et al., 1991; Deshpande et al., 1992), can be synthesized in the vulnerable regions, such as the CA1 region, after ischemia. This view is consistant with the previous study that showed that the neuroprotective actions of basic fibroblast growth factor against excitotoxicity require new protein synthesis (Mattson et al., 1989).
The precise mechanisms underlying neuroprotection by neurotrophic or growth factors against delayed neuronal death remain unknown. Three major hypotheses have been postulated for the occurrence of delayed neuronal death after forebrain ischemia: (1) persisting depression of protein synthesis that precludes long-term survival (Thilmann et al., 1986; Widmann et al., 1991); (2) perturbed signal-transduction pathway that includes imbalance in the activation of growth factors and glutamate receptors (Mattson et al., 1989) and between protein kinase and phosphatases (Saitoh et al., 1991; Wieloch et al., 1993); and (3) sustained perturbation of cell calcium metabolism (Deshpande et al., 1987; Siesjo, 1991). Several lines of evidence have demonstrated that neurotrophic or growth factors antagonize neurotoxicity caused by glutamate (Mattson et al., 1989; Frim et al., 1993) or nitric oxide (Maiese et al., 1993) and reduce neuronal death after hypoxia or glucose-deprivation injury (Cheng and Mattson, 1991). In addition, these factors maintain calcium homeostasis in neurons at risk by inducing calcium-binding proteins, such as calbindin (Cheng and Mattson, 1991; Collazo et al., 1992; Cheng and Mattson, 1994), and reduce oxidant injury by inducing antioxidant enzymes, such as Cu-Zn superoxide dismutase, catalase, and glutathione peroxidase (Pan and Perez-Polo, 1993; Zhang et al., 1993; Goodman and Mattson, 1994; Cheng and Mattson, 1995).
Another important point to consider is whether marked neuroprotective action of PDGF-BB is directly exerted on neurons or indirectly mediated by stimulation of glial cells. Our finding of no significant difference in the number of glial cells on the day of ischemia between PDGF-AA and -BB groups in this study may favor the idea that PDGF-BB exerts its trophic action directly on neurons that possess PDGF β-receptors as described below.
Platelet-derived growth factor-BB was also shown to exert trophic activity on neurons both in vitro and in vivo. Platelet-derived growth factor-BB prolongs the survival and induces neurite outgrowth of cultured rat brain neurons through binding to functional PDGF β-receptors on these cells (Smits et al., 1991). This neurotrophic activity has also been demonstrated on cultured mesencephalic dopaminergic neurons (Nikkhah et al., 1993) and cerebellar γ-aminobutyric acid interneurons (Smits et al., 1993). Recently, it was reported that a brief activation of PDGF-β receptor by PDGF-BB produces a long-lasting inhibition of N-methyl-
In addition, PDGF-BB protects cultured hippocampal neurons against injury caused by glucose deprivation when added before or up to 8 hours after the insult (Cheng and Mattson, 1995). In the case of FeSO4-induced oxidant injury, pretreatment with PDGF-AA or -BB is required to protect the neurons by inducing antioxidant enzymes. This study demonstrated that both PDGF-AA and -BB showed similar neuroprotective actions in each injury paradigm (Cheng and Mattson, 1995). In the current study, however, the potent neuroprotective actions of PDGF-BB against delayed neuronal death were in marked contrast to the lack of significant effects of PDGF-AA. This inconsistency might have been because hippocampal neurons in the adult rat brain express only PDGF-β receptor in vivo, which binds only to PDGF-B chain, although Cheng and Mattson (1995) demonstrated the expression of both PDGF-α and -β receptors in cultured embryonic neurons in vitro. This view is consistent with previous studies that showed that PDGF-α receptor is expressed in glial cells or their precursors and not detected in neurons in embryonic and adult rodents (Pringle et al., 1992; Yeh et al., 1993). In addition, several lines of evidence established ubiquitous neuronal expression of the -β receptors in the brain (Smits et al., 1991; Hutchins and Jefferson, 1992; Iihara et al., 1996a).
Another possible mechanism underlying the neuroprotective effects of PDGF-BB is prevention of the apoptotic component of neuronal death. Recently, increasing evidence has indicated the involvement of apoptotic neuronal death in the injury after both global and focal ischemia (Goto et al., 1990; Shigeno et al., 1990; Papas et al., 1992; Heron et al., 1993; MacManus et al., 1993; Linnik et al., 1993; Okamoto et al., 1993; Roberts-Lewis et al., 1993; Crumrine et al., 1994; Li et al., 1995; MacManus et al., 1995; Du et al., 1996), although some contrasting results have been reported (Deshpande et al., 1992; Dessi et al., 1993), as well as in other injury paradigms, such as neurotrophic factor withdrawal (Martin et al., 1992) and excitotoxicity (Kure et al., 1991). Recently, it was reported that PDGF prevents apoptosis after serum deprivation in PC12 cells expressing the wild-type PDGF receptor through activation of phosphatidylinositol-3 kinase (Yao and Cooper, 1995). Further studies are required to conclude whether PDGF prevents an apoptotic component of ischemic neuronal death after cerebral ischemia.
In conclusion, pretreatment with PDGF-BB, but not PDGF-AA, markedly inhibits delayed neuronal death after forebrain ischemia in rats. In contrast, posttreatment with PDGF-BB showed no significant neuroprotective effect after forebrain ischemia.