
Abstract
Plain language summary
Hepatocellular carcinoma is a highly aggressive cancer. Studies have found that immune cells within tumors—particularly macrophages—play a crucial role in cancer development and therapeutic response. Macrophages have two main “personalities”: the M1 type, which can kill cancer cells, and the M2 type, which supports cancer growth. They shift between these states in response to signals from the tumor microenvironment. Both liver-resident Kupffer cells and infiltrating macrophages are involved in this process. Various molecules, such as cytokines, enzymes, and exosomes, influence their polarization. This polarized state of macrophages, in turn, affects key tumor processes such as stemness, proliferation, metastasis, and angiogenesis. In recent years, scientists have been exploring drugs that specifically target macrophage polarization to suppress liver cancer, offering new directions for future therapy.
Keywords
Introduction
HCC is one of the most prevalent and lethal malignancies worldwide, ranking among the leading causes of cancer-related mortality. Despite advances in diagnostic and therapeutic strategies, most patients are diagnosed at advanced stages, resulting in a poor prognosis and limited treatment options. In recent years, growing evidence has underscored the pivotal role of the TME in shaping HCC initiation, progression, and therapeutic response. Within the TME, macrophages represent key immune regulatory cells that profoundly influence tumor biology. Depending on the surrounding microenvironmental cues, macrophages exhibit remarkable plasticity and can polarize into 2 major functional phenotypes: the classically activated M1 macrophages, which possess pro-inflammatory and antitumor properties, and the alternatively activated M2 macrophages, which promote tumor growth, angiogenesis, and immune evasion. The heterogeneity of hepatic macrophages is particularly notable, encompassing liver-resident Kupffer cells (KCs), infiltrating bone marrow-derived macrophages, and those derived from the peritoneum or spleen. These distinct macrophage populations play dynamic and context-dependent roles in liver injury, fibrosis, inflammation, and tumorigenesis. This review aims to provide a comprehensive overview of the molecular mechanisms governing macrophage polarization and their functional significance in the HCC immune microenvironment. Furthermore, it summarizes recent advances in therapeutic strategies targeting macrophage polarization states, offering insights into potential clinical applications and prognostic implications for improving outcomes in patients with HCC.
Mechanisms Influencing Macrophage Polarization in Liver
Liver macrophages constitute approximately 90% of the body’s macrophage population and exhibit significant heterogeneity, encompassing liver-resident macrophages and various infiltrating macrophage subsets.1,2 Liver-resident macrophages, known as KCs, account for 20-35% of all non-parenchymal liver cells and 80-90% of tissue-resident macrophages in the body. In addition to KCs, liver macrophages include bone marrow/monocyte-derived macrophages, peritoneal macrophages, and spleen-derived macrophages.3,4 Bone marrow-derived monocyte-macrophages are the primary infiltrating macrophage population, recruited following KC and hepatic stellate cell (HSC) activation, and serve as critical contributors to macrophage replenishment and regeneration upon depletion. 5 Peritoneal and spleen-derived macrophages are also recruited to the liver during injury, exerting immunomodulatory functions. 2 In the healthy liver, KCs predominate as sentinel cells, maintaining liver homeostasis. 6 Upon liver injury induced by external factors, KCs are the first to detect signals, differentiating into distinct phenotypes and producing pro-inflammatory or anti-inflammatory factors. Simultaneously, they recruit other macrophages with similar plasticity and diverse functions into the liver.2,5 These macrophages exhibit varied activation states in response to changes in the tissue microenvironment, differentiating into distinct phenotypes under various stimuli, thereby exerting diverse regulatory roles in physiological and pathological processes—a phenomenon termed macrophage polarization. 7
Origin of KCs and Macrophage Polarization
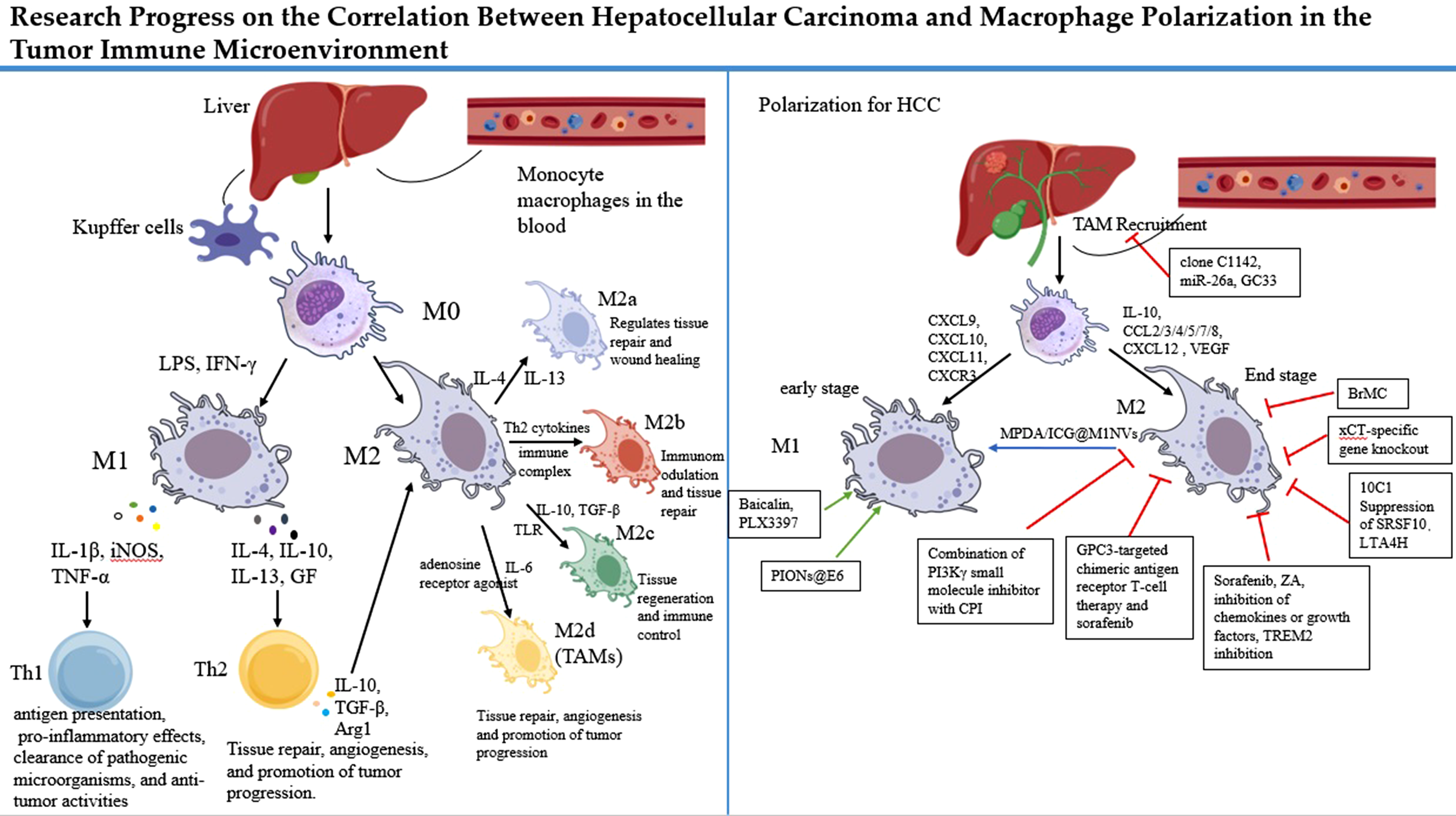
KCs, liver-derived macrophages typically located in hepatic sinusoids, originate from yolk sac-derived progenitor cells seeded in liver tissue during embryonic development.6,8 They can also be replenished through differentiation of bone marrow-derived monocytes. Yolk sac-derived progenitors possess the capacity to differentiate into tissue-resident macrophages while maintaining their population and functionality. Meanwhile, bone marrow-derived macrophages can differentiate into KCs to sustain their numbers, ensuring robust immune defense. KCs self-renew in the liver, remaining quiescent and non-migratory, with functions including pathogen clearance, phagocytosis of cellular debris, and detoxification of gut-derived endotoxins
6
(Figure 1). Origin and Differentiation Pathways of Kupffer Cells.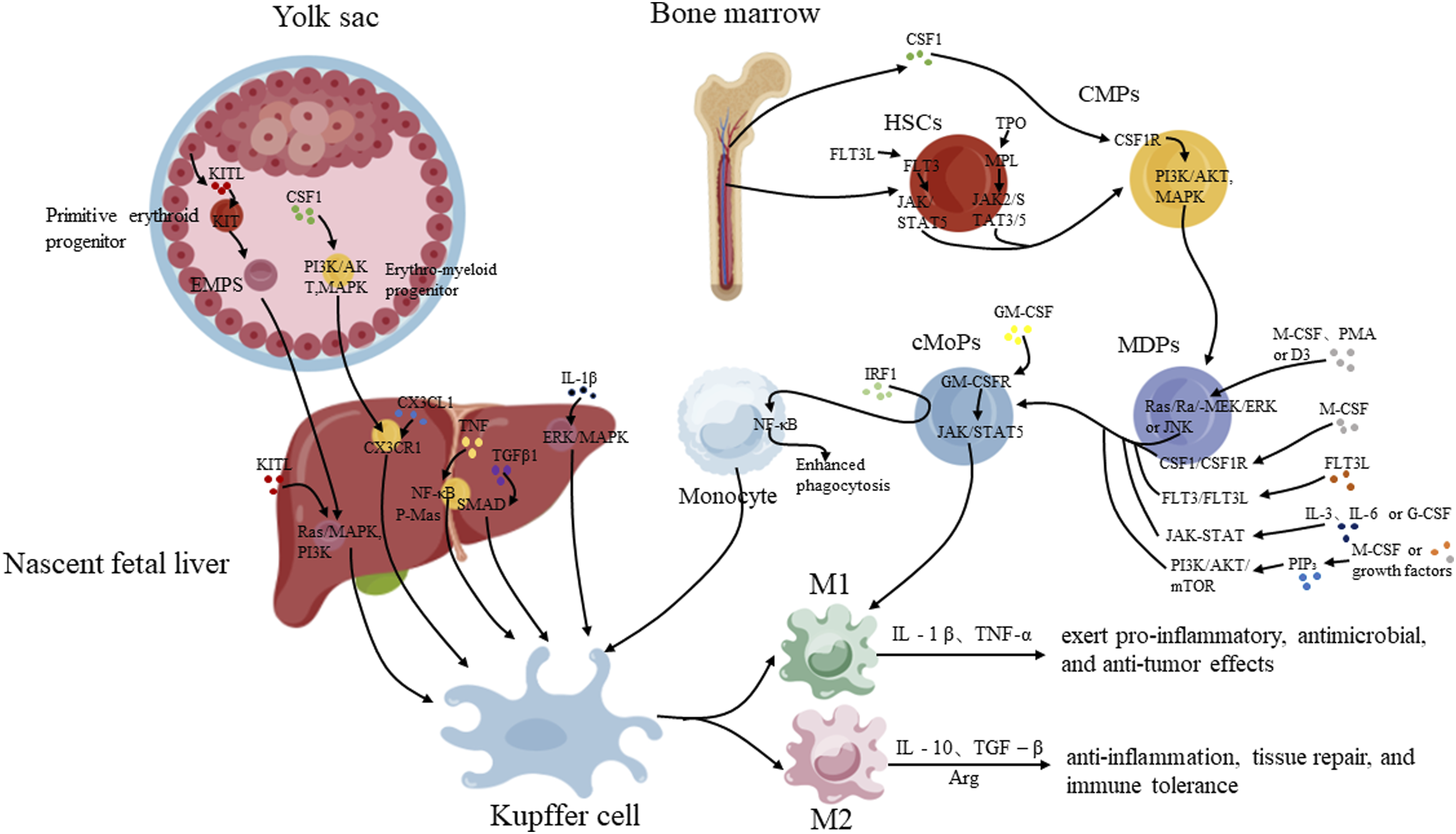
M2 Macrophages can Further Polarize Into Distinct Subtypes Under Specific Inducers, Exhibiting Diverse Functions

Triggering and Activation Mechanisms of KCs
Certain cellular secretions, such as interleukins and tumor necrosis factors, bind to receptors on the cell membrane, activating KCs and modulating signaling pathways to polarize them into M1 or M2 phenotypes. KC activation is triggered by pathogen-associated molecular patterns (PAMPs), cytokines, chemokines, and damage-associated molecular patterns (DAMPs).
Pathogen-associated molecules, primarily bacterial products and toxins like LPS from Gram-negative bacteria, are among the most significant exogenous stimuli for KC activation. LPS activates KCs through 3 main pathways. First, LPS directly activates KCs via the Toll-like receptor (TLR) 4 signaling pathway, upregulating expression of TNF-α, IL-1, IL-6, IL-12, IL-18, IL-10, and IFN-γ. 13 On macrophages, the LPS receptor is a multimolecular complex comprising Cluster of Differentiation (CD) 14, TLR4, and Myeloid Differentiation factor (MD)2. CD14 lacks an intracellular signaling domain, whereas TLR4 contains extracellular and intracellular (Toll/Interleukin-1 Receptor domain [TIR], Toll, and IL-1-related) domains, enabling LPS recognition and intracellular signal transduction, respectively. The soluble accessory molecule MD2 also participates in LPS recognition.14-17 When gut-derived bacteria enter the liver via the portal vein, TLR4 on KCs recognizes LPS, 18 triggering intracellular signaling cascades that activate nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) pathways, inducing pro-inflammatory cytokines (eg, TNF-α, IL-6, IL-1β) and chemokines. Activated TLR4 translocates to an endosomal compartment, recruiting TIR domain-containing adaptor proteins, inducing TIR-domain-containing adapter-inducing interferon-β (TRIF), TRIF-related adaptor molecule (TRAM), TNF receptor-associated factor 3 (TRAF3), and other proteins, leading to phosphorylation of interferon regulatory factor 3 (IRF3) and type I interferon (IFN) responses. 8 Second, LPS activates KCs by binding to endosomal TLR4, inducing TRIF complex formation, which triggers type I IFN responses and pro-caspase-11 expression. Finally, LPS can be sensed in the cytoplasm by directly binding caspase-11 in a TLR4-independent manner, 19 leading to caspase-11 oligomerization and auto-activation. Caspase-11, a direct cytoplasmic LPS receptor, is activated through oligomerization upon LPS interaction, 20 cleaving the precursor form of Gasdermin D (Gsdmd) to produce an N-terminal fragment, triggering pyroptosis (inflammatory cell death) and NOD-like receptor family, pyrin domain containing 3 (NLRP3)-dependent caspase-1 activation, critical for cytokine processing (IL-1β and IL-18).20-23 Recent studies have identified drugs that suppress M1 macrophage polarization by inhibiting the TLR4/NF-κB pathway. For instance, berberine competitively inhibits the binding of TLR4 and Myeloid differentiation primary response 88 (MyD88) Inhibits the TLR4/MyD88/NF-kB signaling pathway and thus inhibits M1 macrophage polarization. 24 Quercetin downregulates NF-κB and IRF5 expression, inhibiting upstream TLR4/MyD88 activity and M1 polarization.25,26
The cytokine IFN-γ binds its receptor and activates JAK, inducing STAT1 phosphorylation and driving M1 polarization.27,28 IFN-γ also promotes the metabolism of M1 macrophages, thereby enhancing their cell viability and pro-inflammatory activity via the Janus kinase/Signal transducer and activator of transcription (JAK/STAT)1 pathway
29
(Figure 2). Additionally, IFN-γ sensitizes macrophages to inflammatory mediators and synergizes by blocking feedback inhibition of TLR signaling, while NF-κB and MAPK enhance JAK/STAT1 transcriptional activity.
27
IL-4 inhibits M1 and induces M2 polarization through the JAK/STAT6 pathway.
30
Curcumin upregulates STAT6 expression by secreting IL-4 and IL-13, promoting M0 and M1 macrophage polarization toward M2.31,32 Growth differentiation factor 3 from the TGF-β superfamily inhibits M1 and promotes M2 polarization by enhancing Smad2 and Smad3 phosphorylation.
33
The atypical chemokine CX3CL1 (fractalkine) promotes liver dendritic cell maturation and survival of infiltrating macrophages.34,35 Key chemokine pathways for inflammatory monocytes include CC chemokine ligand (CCL)2-CC motif chemokine receptor (CCR)2, CCL1-CCR8, CCL5-CCR1/CCR5, and C-X-C motif chemokine ligand (CXCL)10- C-X-C motif chemokine receptor (CXCR)3 interactions36,37 (Figure 2). Signaling pathways governing macrophage polarization and survival.
Macrophage polarization is a complex process modulated by the surrounding immune microenvironment. Hepatic macrophages, primarily comprising KCs, under-go activation and polarization. Under pathological conditions, macrophage recruitment occurs, involving not only resident KCs but also circulating monocyte-derived macrophages. These cells polarize in response to cytokines, specific enzymes, exosomes, or interactions with other cells, thereby mediating immune responses.
Molecular Mechanisms of Macrophage Polarization in HCC
During tumor development, tumor-associated antigens and tumor-specific antigens are highly expressed in cancer cells, activating dendritic cells, initiating antigen presentation, and stimulating lymphocytes to exert anti-tumor activity. However, as tumor cells counteract the immune system, an immunosuppressive microenvironment forms, triggering a series of immune escape mechanisms. These mechanisms allow tumor cells to evade immune attacks, leading to infiltration or distant metastasis. This includes recruiting immunosuppressive cells and inducing T-cell exhaustion to reduce anti-tumor immune responses. 38 The TME consists of tumor cells, immune cells (eg, TAMs, T and B lymphocytes, NK cells), stromal cells, fibroblasts, pericytes, and non-cellular components (eg, extracellular matrix [ECM]). 39 The TME plays a critical role in cancer growth, influencing disease prognosis and immune responses. 40
TAMs are the most abundant immune cells in the TME. They promote angiogenesis, ECM degradation, and remodeling, as well as tumor cell motility, facilitating immune escape. 41 TAMs differentiate from macrophages under the influence of various factors in the TME, adopting the phenotype and function of M2-type macrophages. 42 These cells secrete multiple cytokines that promote tumor progression. Numerous studies indicate that high TAM density in malignant tumors, such as thyroid cancer, 43 pancreatic cancer,44,45 and ovarian cancer, 46 is closely associated with tumor invasion and reduced survival rates. In the early stages of HCC, a fibrotic and pro-inflammatory microenvironment is characterized by activated HSCs and M1-polarized macrophages, which drive ECM deposition and inflammatory signaling, supporting tumorigenesis. 47 In contrast, in advanced HCC, the TME is dominated by immunosuppressive M2-type TAMs, which polarize under the influence of cytokines such as TGF-β, IL-10, and IL-13. 48 These M2 TAMs promote tumor progression by suppressing effector T cells, enhancing angiogenesis, and facilitating metastasis. 48 Targeted therapies against TAMs primarily involve reprogramming TAMs toward an M1 phenotype, eliminating existing TAMs, and limiting the recruitment of monocytes and M2 macrophages. This chapter focuses on the molecular mechanisms influencing macrophage polarization in the HCC immune microenvironment, reviewing the effects of cytokines, exosomes, and direct interactions secreted by HCC cells and immune cells on macrophage polarization. Identifying new therapeutic targets will aid in the diagnosis and treatment of HCC, improving prognosis.
HCC Influences Macrophage Polarization
Macrophage polarization plays a critical role in the occurrence, progression, and treatment resistance of HCC. Macrophages can be induced by the microenvironment to polarize into pro-inflammatory or anti-inflammatory/pro-tumor states. In HCC, tumor cell-secreted cytokines, exosomes, and enzymes stimulate macrophages to polarize into the M2 type, thereby promoting tumor progression.
Cytokines
HCC cells secrete cytokines that stimulate the monocyte-macrophage system, recruiting large numbers of monocytes and unpolarized macrophages from the blood into the TME. These cells are then polarized into M1 or M2 macrophages under the influence of TME factors, exerting distinct roles. Cytokines and molecules in the TME, such as IFN-γ, TNF-α, monoglyceride lipase, and CD68, induce M1-TAM polarization, which exhibits anti-tumor activity 49 . Hypoxia-inducible factors, peripheral cannabinoid receptor type 2 (CB2) receptors, CD163, signal transducer and STAT, and arginase are primarily responsible for M2-TAM polarization and tumor progression. 50 M1-TAMs suppress tumors, while M2-TAMs promote tumor development. Chemokines such as CXCL9, CXCL10, CXCL11, and CXCR3 51 drive macrophage polarization toward the M1 phenotype (Figure.3). IL-12 induces M1-like macrophage polarization by downregulating Stat-3, inhibiting HCC growth 52 . The IL-6/STAT3 signaling pathway is suppressed in M1 macrophages but activated in M2 macrophages, promoting HCC cell viability, proliferation, invasion, migration, and drug resistance 53 . Anti-IL-6 treatment inhibits this pathway, promoting M1 polarization and anti-tumor effects.
Chemokines such as CXCL9, CXCL10, CXCL11, and CXCR3
51
drive macrophage polarization toward the M1 phenotype (Figure 3). IL-12 induces M1-like macrophage polarization by downregulating Stat-3, inhibiting HCC growth.
52
The IL-6/STAT3 signaling pathway is suppressed in M1 macrophages but activated in M2 macrophages, promoting HCC cell viability, proliferation, invasion, migration, and drug resistance.
53
Anti-IL-6 treatment inhibits this pathway, promoting M1 polarization and anti-tumor effects. Molecular interactions between immune cells, macrophages, and TME in cancer progression.
Cytokines such as IL-10, CCL2/3/4/5/7/8, CXCL12, and VEGF 54 drive macrophage polarization toward the M2 phenotype (Figure 3). IL-1β mediates immunosuppression by upregulating programmed death-ligand 1 (PD-L1) expression in tumor cells. 55 In the TME, macrophage colony-stimulating factor (M-CSF) and CCL-2 are highly overexpressed, increasing the proportion of TAMs.56,57 Chemokines such as CCL, vascular endothelial growth factor (VEGF), and platelet-derived growth factor (PDGF) facilitate the recruitment of M2 macrophages to the TME58,59 (Figure 3). Additionally, studies have shown that Wnt10a regulates IL-3 secretion via the ERK5/1-STAT2 pathway, inducing M2 polarization in colorectal cancer (CRC). 60
Enzymes
Various enzymes in the TME regulate macrophage polarization through metabolic regulation, signal transduction, epigenetic modifications, and proteolysis, influencing tumor immune escape, invasion, and metastasis. These enzymes are critical targets for tumor therapy research.
Reactive oxygen species (ROS) play a pivotal role in the TME and are involved in regulating macrophage polarization. ROS accumulation promotes the polarization of TAMs toward the M2 phenotype, which is immunosuppressive and supports tumor growth and immune escape.61-63 Thus, regulating ROS levels is an important strategy for modulating the immune microenvironment and inhibiting tumor progression. In HCC, ROS are primarily generated via NADPH oxidase 2 (NOX2). NOX, a membrane-bound enzyme widely expressed in phagocytes (also known as NOX2), produces ROS to combat pathogens by regulating inflammasomes, autophagy, and type 1 IFN signaling. 64 Beyond pathogen defense, NOX is implicated in the polarization and differentiation of tumor-associated immune cells, including myeloid-derived suppressor cells and TAMs.65-67 Studies show that HCC-derived high-mobility group box 1 (HMGB1) stimulates NOX2-dependent ROS production via TLR2, triggering autophagy and leading to lysosomal degradation of NF-κB p65, thereby maintaining M2 macrophage polarization 68 (Figure 3). Notably, interrupting this pathway significantly alters the tumor immune microenvironment. Specifically, blocking HMGB1 signaling or clearing ROS reduces M2-TAM accumulation and effectively inhibits HCC growth in mouse models. 68 This study reveals a novel regulatory mechanism of the HMGB1-TLR2/NOX2/autophagy axis in HCC-induced M2 macrophage polarization.
Matrix metalloproteinases (MMPs) are a group of proteolytic enzymes critical for ECM remodeling. The positive interaction between MMP-8 and TGF-β1 maintains the M2 macrophage phenotype, promoting HCC invasion and metastasis. 69 Long non-coding RNA (lncRNA) P73 antisense RNA 1T (TP73-AS1) has been identified in various cancers, including HCC, 70 and is implicated in promoting cancer cell proliferation and tumorigenesis. 71 Bioinformatics analysis revealed potential interactions between TP73-AS1 and miR-539, predicting MMP-8 as a potential target of miR-539. 72 The TP73-AS1/miR-539/MMP-8 axis regulates M2 macrophage polarization in HCC via the TGF-β1 signaling pathway. 73 TP73-AS1 and MMP-8 levels are significantly elevated in HCC tumor tissues, with TP73-AS1 regulating MMP-8 expression by targeting miR-539, and MMP-8 modulating M2 macrophage polarization through TGF-β1 signaling. Knockdown of TP73-AS1 and overexpression of miR-539 inhibit HCC tumor growth and M2 macrophage infiltration. 73
The Notch signaling pathway, highly conserved in evolution, regulates cell proliferation, differentiation, and apoptosis, significantly influencing liver vasculature, bile duct, and immune cell74,75 development. Macrophages stably express Notch ligands and receptors (Notch1, 2, and 4), suggesting Notch signaling’s role in macrophage activation. 76 When Notch signaling is activated, secretion of inflammatory cytokines IL-6 and iNOS increases, IL-10 release decreases, and macrophages polarize toward the M1 phenotype. 77 Studies indicate that RBP-J-mediated Notch signaling and TLR signaling synergistically regulate macrophage function, with Notch signaling effects mediated by feedback regulation of downstream genes Hes1 and Hey1. 78 Suppressor of cytokine signaling 3 (SOCS3) is a downstream molecule of the Notch signaling pathway, regulating M1 and M2 macrophage polarization via RBP-J-mediated Notch signaling. 79 Changes in macrophage phenotype mediated by Notch signaling play a significant role in liver tumor development. 80 In HCC, activating Notch signaling promotes M1 macrophage differentiation, enhancing inflammation and anti-tumor activity, while blocking Notch signaling polarizes macrophages toward M2, suppressing inflammation and promoting tumor growth. 81
Exosomes
Exosomes, secreted by cancer or stromal cells, release proteins, microRNAs, cytokines, and other molecules to regulate macrophage polarization (Figure 3).
HCC cell-derived miR-200b-3p exosomes are internalized by M0 macrophages, downregulating ZEB1 and upregulating IL-4 to induce M2 polarization. The JAK/STAT signaling pathway is activated in M2 macrophages, increasing Proto-oncogene serine/threonine-protein kinase Pim-1 (PIM1) and VEGFA expression. These cytokines accelerate HCC proliferation and metastasis, forming a feedback loop between HCC cells and M2 macrophages. 82 Inhibiting or blocking miR-200b-3p exosomes reduces M2 macrophage polarization, suppressing HCC proliferation and metastasis. Additionally, targeting key proteins in this pathway is critical for HCC treatment.
Proteasome subunit alpha 5 (PSMA5) is an HCC cell-secreted exosomal component associated with patient survival. Exosomes isolated from HCC cells by Xiao Zuo were successfully internalized by macrophages, promoting M2 polarization and JAK2/STAT3 pathway activation. Knockdown of PSMA5 in HCC cell-derived exosomes inhibited their induction of macrophages, reducing the promotion of HCC cell migration/invasion, tumorigenesis, and in vivo M2 polarization and JAK2/STAT3 pathway activation. 83 Developing PSMA5 inhibitors could reduce HCC invasion and metastasis.
Small nucleolar RNAs (snoRNAs), located in the nucleolus, are a large family of endogenous non-coding RNAs, ranging from 60 to 140 nucleotides. 84 Based on structural features, conserved sequences, and small nucleolar ribonucleoproteins formed, snoRNAs are classified into H/ACA box snoRNAs and C/D box snoRNAs. 85 SnoRNAs are involved in post-transcriptional modification of ribosomal RNA (rRNA), and defects in ribosome maturation and function may lead to the transformation of normal cells into pathological cells. Studies have found that SNORD52 is enriched in HCC cell-derived exosomes and plasma samples from HCC patients, inducing M2 macrophage polarization by activating the JAK2/STAT6 pathway. 86
Exosomes (H-Exo and N-Exo) derived from HCC promote M2 macrophage polarization, accelerating HCC cell migration and epithelial-mesenchymal transition (EMT). In the HCC TME, the miR-130b-3p-PTEN-PI3K-Akt signaling pathway induces M2 macrophage polarization. In vivo, oleic acid (OA) treatment enhances the efficacy of anti-PD1 antibodies by reducing M2 macrophage numbers and increasing CD8+ T cell counts. 87
Immune Cells Influence Polarization
Immune cells profoundly influence macrophage polarization through direct cell-cell interactions or secretion of specific cytokines and chemokines. The following sections detail the impact of different immune cells on macrophage polarization (Figure 3).
T Cells and Macrophage Polarization
T cells play a critical role in immune regulation by influencing macrophage polarization toward M1 or M2 phenotypes through cytokine secretion, surface molecule interactions, and metabolic regulation, impacting tumor immunity (Figure 3).
Th1 cell-secreted cytokines, such as IFN-γ and TNF-α, are key inducers of M1 macrophage polarization. M1 macrophages exhibit pro-inflammatory and tumor-suppressive functions, secreting pro-inflammatory cytokines and chemokines to attract and activate other immune cells to combat tumors. TNF-α enhances macrophage pathogen- and tumor-killing capabilities, reinforcing the M1 phenotype. IFN-γ induces rapid activation of aerobic glycolysis in macrophages, sustaining the IFN-γ-triggered JAK/STAT1 signaling pathway and promoting in vitro M1 polarization. 27 The M1 state, induced by IFN-γ and microbial ligands via TLR signaling, is primarily controlled by STAT1,88,89 IRF190,91 and NF-κB, 92 while the M2 state, induced by IL-4, IL-10, and IL-13, is regulated by STAT3, 93 STAT6, 94 and PPARγ. 95
M2 polarization is influenced by Th2 cytokines, such as IL-4, IL-6, IL-10, IL-13, TGF-β, glucocorticoids, immune complexes, TLR or IL-1R ligands, and leukemia inhibitory factor. 96 Tumor-site T cells (ie, Th2 cells) and cancer cells coordinate an M2-like microenvironment and TAM differentiation. 97 Activation of the M2/Th2 response determines the release of growth factors and cytokines (eg, IL-4, IL-10, IL-13, and TGF-β). 98 IL-4 and IL-13 induce M0 macrophage polarization into M2 macrophages, 99 enhancing HCC cell migration. In gastric cancer, IL-4 induces metabolic changes in macrophages, activating the PI3K/AKT/mTOR pathway, promoting ATP production, enhancing glycolysis, increasing lactate production, and upregulating FcγRIIB expression, leading to CD8+ T cell dysfunction and resistance to anti-PD-1 therapy. 100 Studies have shown that IL-4/STAT6 signaling activation promotes lung cancer progression by increasing M2 myeloid cells. 101 Thus, IL-4 is a key stimulator of M2 macrophage polarization. Deng et al demonstrated that Phorbol 12-myristate 13-acetate (PMA)-IL-4 stimulation of U937 cells induces monocyte differentiation and M2 macrophage polarization, 102 confirming IL-4’s role in polarizing M0 macrophages into M2.
Treg cells secrete IL-10 and TGF-β, suppressing macrophage pro-inflammatory functions and promoting M2 polarization. Tumor-derived Tregs regulate TAM metabolic adaptation by suppressing CD8 T cell IFN-γ production and promoting M2 TAM conversion. 103 Studies show that focal adhesion kinase (FAK) promotes HCC by increasing Treg cells, polarizing macrophages from M1 to M2 via the PI3K/AKT/JAK/STAT2 and p38/JNK pathways. 104 TGF-β recruits M2 macrophages in HCC, which are further polarized by connective tissue growth factor (CTGF). TGF-β acts as a chemoattractant, recruiting monocytes from peripheral blood, while CTGF transforms monocytes into M2 macrophages, stimulating tumor growth and secreting CCL18 to promote HCC cell migration.105,106 IKAROS family zinc finger 1 (IKZF1), a critical transcription factor in lymphocyte development, is associated with immune regulation disorders, particularly affecting late T/B cell differentiation. IKZF1 interacts with Foxp3, suppressing gene expression in Treg cells and regulating autoimmune and anti-tumor immunity. 107 In gliomas, CCL2 promotes IKZF1 expression, inducing M2 polarization via the CD84-SHP2 signaling axis. 108 Additionally, the IKZF1-IRF4/IRF5 axis regulates macrophage polarization in multiple myeloma. 109 Liu et al found that IKZF1 induces pyroptosis and inhibits M2 macrophage polarization by suppressing the JAK2/STAT5 signaling pathway, 110 inhibiting colon cancer metastasis, providing a theoretical basis for colon cancer treatment.
TAMs and Neutrophils
Changes in TAMs also affect neutrophil differentiation. Similar to M1 and M2 macrophages, neutrophils can differentiate into N1 (anti-tumor) and N2 (pro-tumor) phenotypes based on their functions. Tumor cells and T cells secrete TGF-β, which promotes tumor-associated neutrophil (TAN) differentiation into the pro-tumor N2 phenotype, while IFN-β induces TANs to adopt the anti-tumor N1 phenotype.111,112 TANs secrete CCL2 and CCL17 to recruit macrophages and Tregs, promoting tumor invasion and angiogenesis. 113 The pro-inflammatory cytokine IL-17 facilitates TAN recruitment to the TME. CCL2 and CCL17, strongly produced by TANs and peripheral blood neutrophils, recruit macrophages to the TME 114 (Figure 3). TAN infiltration positively correlates with angiogenesis at the tumor invasion margins in HCC patients. 115
TAMs and Cancer-Associated Fibroblasts (CAFs)
CAFs are key TME components, promoting tumor invasion, metastasis, and drug resistance through ECM remodeling, cytokine secretion, and communication with various cells.116,117 CAF-secreted IL-8 promotes TAM recruitment and alters the TME. 118 In HCC, CAFs are recruited and adhere to macrophages, secreting Growth Arrest–Specific 6 (GAS6) to promote M2 macrophage polarization (Figure 3). Injecting human antibody IgG78 into tumor-bearing mice specifically attenuates the effects of endocrine factors on CAF-macrophage interactions, slowing tumor growth. 119
Advances in Macrophage Polarization Based on Single-Cell Sequencing, Multi-Omics and ST
With the rapid advancement of single-cell RNA sequencing (scRNA-seq), multi-omics, and ST technologies, researchers are now able to delineate the polarization spectrum and functional states of tumor-associated macrophages with much greater precision across multiple dimensions, including single-cell heterogeneity, molecular mechanisms, and spatial organization. In this section, we summarize recent advances in TAM polarization revealed by these cutting-edge approaches and discuss their functional implications and therapeutic potential.
Application of scRNA-seq Technology in the Study of Macrophage Polarization
ScRNA-seq provides single-cell resolution and enables an in-depth dissection of the heterogeneity and functional states of cellular populations within the TME, offering a new perspective for studying macrophage polarization. Early large-scale single-cell studies have demonstrated that the myeloid compartment in the HCC TME is highly heterogeneous, comprising liver-resident Kupffer cells, monocyte-derived macrophages, as well as diverse neutrophil and dendritic cell subsets. This complexity goes far beyond the traditional and overly simplistic “M1/M2” dichotomy.
Building on these findings, recent integrative single-cell studies have further refined the macrophage landscape in HCC. For example, one study integrated 4 publicly available HCC scRNA-seq cohorts (GSE140228, GSE125449, GSE149614, and GSE156625) and identified 6 subsets of monocyte-derived macrophages (Macro1–Macro6) and 4 subsets of Kupffer cells (Kupffer1–Kupffer4). Among them, CXCL10+ pro-inflammatory–like macrophages and MT1G+ Kupffer cells are predominantly enriched in tumor tissues and display enhanced antigen-presenting capacity and inflammatory signaling. In contrast, another subset of TAMs is characterized by high expression of immunosuppressive markers such as MRC1/CD163 and TREM2, and functionally exhibits an M2-like, immunoregulatory phenotype. 120 Multiple single-cell “ecosystem” atlases of HCC, including cohorts of early recurrent and metastatic disease, consistently highlight SPP1+ (osteopontin+) macrophages as a recurrent population across virtually all datasets. These SPP1+ TAMs frequently co-occur with features of angiogenesis, extracellular matrix remodeling, and T-cell exhaustion, and are therefore considered a highly robust signature of immunosuppressive TAMs. 121
Single-cell pseudotime analyses combined with gene regulatory network (GRN) inference indicate that macrophage polarization in HCC is not a binary, switch-like event, but rather a continuous reprogramming process along defined trajectory. Starting from a population of pro-inflammatory–like macrophages characterized by high expression of inflammatory cytokines and chemokines (eg, CXCL9/10, TNF, IL1B), cells progressively acquire an anti-inflammatory and immunoregulatory phenotype along the pseudotime axis, marked by upregulation of anti-inflammatory mediators and immune checkpoint molecules (such as IL10, TGFB1, PD-L1, and VSIR) and downregulation of antigen-presentation molecules, ultimately converging toward an immunosuppressive TAM subset with high expression of MRC1/CD163/TREM2. 120 Transcription factor enrichment analyses further suggest that NF-κB, STAT1, and IRF5 predominantly drive the early pro-inflammatory/anti-tumor state, whereas STAT3, PPARγ, and MAF gradually become dominant at later stages, promoting M2-like polarization and the acquisition of tissue-repair and pro-tumorigenic phenotypes. 48
Re-analysis of public HCC single-cell datasets has revealed a distinct SPP1+BCL2A1+ double-positive subset within SPP1+ TAMs. This subset displays a combination of strong anti-apoptotic, pro-inflammatory, and immunosuppressive features, with a transcriptional program closely associated with angiogenesis, EMT, and tumor stemness. Notably, a higher proportion of this SPP1+BCL2A1+ TAM subset correlates with worse overall survival. 122 Cell–cell communication analyses further demonstrate that macrophages and tumor cells are interconnected through multiple enriched ligand–receptor axes, including SPP1–CD44, CCL2–CCR2, and CXCL12–CXCR4. 121 These signaling pathways not only drive macrophages toward an M2-like immunosuppressive state, but also reciprocally enhance tumor cell invasion and maintenance of stem-like properties.
Single-cell studies linking macrophage polarization to clinical phenotypes and responses to immunotherapy have also made notable progress. Across multiple cohorts, enrichment of SPP1+ or SPP1+BCL2A1+ TAMs are significantly associated with vascular invasion, advanced tumor stage, and poor prognosis, and frequently co-occurs with accumulation of CAFs, collagen deposition, and upregulation of angiogenesis-related gene programs, together constituting key components of an “invasive niche”. 122 In single-cell analyses of HCC patients treated with immune checkpoint inhibitors (ICIs), non-responding tumors are often enriched in SPP1+, TREM2+, and CD163+ TAMs and are accompanied by a profoundly exhausted CD8+ T-cell phenotype (co-expression of PD-1, LAG3, and TIM-3) and reduced T-cell clonal diversity. 123 Mechanistic studies further indicate that several molecules, such as GSDME and ANGPTL4, can modulate the efficacy of PD-1 blockade by regulating the balance between inflammatory and M2-polarized states of macrophages, highlighting macrophage polarization as a promising therapeutic target to overcome resistance and improve clinical benefit from immunotherapy. 124
Despite the unprecedented resolution provided by scRNA-seq for profiling the transcriptional and functional heterogeneity of macrophages, several limitations remain, including inter-patient variability, sampling heterogeneity, batch effects, and discrepancies between analytical pipelines. Future studies will need to integrate single-cell omics with functional assays, ST, metabolomics, and other multi-omics approaches to systematically validate the functional roles of distinct macrophage subsets and elucidate their mechanistic contributions to HCC initiation, progression, and responses to immunotherapy.
Macrophage Polarization in the Multi-Omics Context
Multi-omics approaches, which integrate genomic, transcriptomic, epigenomic, proteomic, and metabolomic data, offer a comprehensive view of the TME. In HCC, the multi-dimensional mechanisms underlying macrophage polarization are gradually being elucidated through multi-omics studies.
The core value of multi-omics analysis lies in projecting macrophage subsets identified at the single-cell level to large cohorts (eg, TCGA/GEO), quantifying their impact on prognosis and drug response. One study first identified SPP1+ TAMs in HCC single-cell data, and then used Mendelian randomization combined with LASSO regression to select 16 robust SPP1+ TAM-related genes from bulk transcriptomic data in TCGA/GEO, constructing a SPP1+ TAM Risk Score (STRS). 125 Patients with high STRS not only exhibited significantly worse overall survival but also displayed an immune-suppressive TME characterized by a notable increase in Tregs, M2-like TAMs, and exhausted CD8+ T cells. Additionally, stronger activation of TGF-β, IL-10, and Wnt signaling pathways, as well as a potential resistance to immune checkpoint inhibitors, were observed. A 2025 study by MDPI Diagnostics confirmed the presence of SPP1+BCL2A1+ TAMs in public HCC single-cell datasets and established corresponding risk signatures based on TCGA/GEO, further linking this subset with early recurrence, vascular invasion, and immune rejection. 122
HCC is characterized by significant metabolic reprogramming, and multi-omics studies are increasingly linking metabolic subtypes to immune infiltration patterns, particularly TAM polarization. One integrated study combining single-cell and bulk transcriptomic data classified HCC into glycan-HCC (glycosylation-dominant) and lipid-HCC (lipid metabolism-dominant) subtypes. The former enriched glycosylation, ECM remodeling, and immunosuppressive pathways, along with an increase in M2-like TAMs and Tregs, correlating with poor prognosis. 126 A recent multi-omics study published in Cancers analyzed the transcriptome, methylome, and metabolome of 60 HCC patients, creating an immune activity score and categorizing patients into high and low immune activity groups. In the high immune activity group, despite the presence of active T cells and NK cells, lipid metabolic reprogramming and the accumulation of suppressive TAMs were observed in some samples, suggesting that the metabolic environment may influence macrophage polarization and regulate the quality of anti-tumor immunity. 127 Non-coding RNAs, especially lncRNAs, play a critical “hub” role in connecting multi-omics signals to cellular polarization outcomes. A 2024-2025 review systematically summarized how key lncRNAs, including Highly Upregulated in Liver Cancer (HULC), Metastasis-Associated Lung Adenocarcinoma Transcript 1 (MALAT1), miR222/221 cluster host gene (MIR222HG), and Maternally Expressed Gene 3 (MEG3), shape macrophage polarization in HCC by regulating cytokines such as IL-10, TGF-β, and CSF1, as well as epigenetic modifications such as m6A and methylation: HULC and MALAT1 typically upregulate immunosuppressive factors through ceRNA mechanisms, promoting M2-like polarization of TAMs; MIR222HG enhances the tumor cells’ induction of macrophage polarization via the LIN28B/ATG5 axis; whereas MEG3 has been reported to suppress M1 polarization and mitigate inflammatory responses, indirectly affecting tumor immunity. 128 Studies employing DNA methylation, histone modifications, and other multi-omics techniques have found that HCC subtypes associated with the accumulation of immunosuppressive TAMs often exhibit hypermethylation of upstream promoters in the IL-10/TGF-β axis and epigenetic inactivation of genes involved in lipid metabolism, suggesting that epigenetic reprogramming could provide a stable foundation for maintaining M2-like polarization. 129
The integration of multi-omics data with spatiotemporal evolution and immunotherapy prediction is an ongoing area of development. A 2023 report in Cancer Cell presented a multi-dimensional atlas combining genomics, transcriptomics, single-cell sequencing, ST, and immunohistochemistry of 182 HCC patients. This study revealed a gradual enrichment of immune-suppressive myeloid cells (including SPP1+ TAMs) alongside tumor cell clonal evolution, from primary to metastatic sites, suggesting that macrophage polarization plays a critical role in driving immune escape and therapeutic resistance in a spatiotemporal manner. 130 Another multi-omics study integrating bulk RNA, proteomics, single-cell, and spatial data constructed a Tumor Purity (TP)-TME prognostic model. The results showed that in the high TME risk group, TAM markers such as SPP1/CD163/MRC1, as well as CAF markers, were significantly elevated and correlated with resistance to immune checkpoint inhibitors. 131
While multi-omics analysis has provided multidimensional insights into macrophage polarization, integrating different types of data still faces technical and biological challenges. The heterogeneity of data, batch effects, and the complexity of biological contexts necessitate the development of advanced algorithms and validation experiments.
Advances in Macrophage Polarization in HCC Based on ST
In HCC, traditional studies on macrophage polarization and functional heterogeneity primarily rely on tissue homogenate analysis, immunohistochemistry, and flow cytometry, which are mainly based on surface markers for identifying macrophage subsets. However, these methods often overlook the crucial spatial dimension of cells within tissues. ST integrates transcriptomic data with precise spatial coordinates, preserving the in situ structure of tissue slices, and helps to unveil the distribution patterns and functional states of immune cells in different tissue microenvironments.
ST allows researchers to map the spatial distribution of immune cells and their polarization states while maintaining the tissue morphology. One study obtained 7553 ST spots from HCC samples and identified 15 spatial subtypes through clustering. It was found that the tumor core exhibited a high expression of CCL15, along with enrichment of M2 markers such as CD163 and Mannose Receptor C-Type 1 (MRC1), and immune-suppressive pathways. This region correlated with the worst prognosis. In contrast, the tumor margin and certain stromal regions exhibited high expression of CCL19/CCL21 and were enriched with CD3+ T cells and CD20+ B cell infiltration, correlating with significantly better prognosis. 132 Combined ST and functional experiments revealed that the tumor immune barrier (TIB) at the tumor margin, consisting of SPP1+ macrophages and FAP+ CAFs, is a critical spatial structure determining the efficacy of immune checkpoint inhibitors. Blocking SPP1 can disrupt the TIB, promoting CD8+ T cell infiltration into the tumor core, thereby significantly improving the efficacy of anti-PD-1 therapy. 133 ST further suggested that tumor-derived SPP1 binds to the CD44 receptor to induce HSC differentiation into CAFs, forming a spatial niche in the tumor margin and fibrotic areas where SPP1+ tumor cells, CAFs, and M2-like macrophages co-localize. This niche not only strengthens the matrix barrier but also exacerbates local immune suppression. 134
Recent spatial immunology studies have begun to focus on “how macrophages are spatially organized around T cells and how this arrangement affects Cytotoxic T Lymphocyte (CTL) function.” Researchers performed single-cell and spatial multiplex staining analyses on 20 cases of HCC with high CTL infiltration, and validated the findings in 386 independent samples. They found that when macrophages form dense clusters in spatial proximity to CTLs, they typically exhibit lipid metabolism-driven M2-like polarization, accompanied by CTL functional exhaustion and poor prognosis. In contrast, when macrophages are more diffusely distributed, CTLs maintain better effector functions, suggesting that the spatial organization of macrophages is a key factor influencing the quality of anti-tumor immunity. 135 Single-cell and spatial multi-omics analyses further revealed that certain highly invasive tumor cell subsets in HCC are spatially co-localized with F5-CAFs and SPP1+ TAMs, forming a “malignant ecosystem” that promotes tumor stemness maintenance and immune escape. This highlights the spatial coupling relationship between macrophage polarization, CAFs, and tumor cell clonal evolution. 136
Despite the significant advancements ST has made in understanding the spatial mechanisms of macrophage polarization, technical limitations remain, including restrictions on spatial resolution, insufficient probe or gene coverage, and challenges in deconvoluting and accurately defining macrophage fine subsets. Future studies could combine ST with functional validation techniques, such as in situ perturbation, macrophage function assays, and metabolomics/lipidomics analysis, complementing them with single-cell multi-omics and spatial proteomics, to elucidate the pathogenic roles more systematically and druggable targets of macrophages in the HCC microenvironment.
Overall, single-cell transcriptomics has revealed the high heterogeneity of TAMs in HCC at the level of lineage origin, transcriptional characteristics, and polarization trajectories, identifying multiple functional subsets such as SPP1+, TREM2+, and CXCL10+ along with their regulatory networks. Multi-omics studies have integrated these macrophage subsets with genomic, epigenomic, and metabolomic information, constructing prognostic models and molecular subtypes represented by SPP1+ TAMs and emphasizing the significant role of metabolic reprogramming and lncRNAs in macrophage polarization. ST and spatial multi-omics have further uncovered key spatial structures such as the CCL15–CD163 region, TIB, and the co-localization of SPP1+ TAM–CAF–tumor cells, directly linking macrophage polarization patterns with CTL function, immune rejection, and responses to immunotherapy. The integration of these 3 approaches provides a multi-scale framework for macrophage polarization research from molecules to cells, tissues, and patients, laying a solid foundation for the development of precision therapeutic strategies targeting TAM polarization and remodeling the tumor immune microenvironment.
Impact of Macrophage Polarization on HCC Progression
TAMs, as the most abundant immune cells in the tumor immune microenvironment, exert a significant influence on the progression of HCC, primarily through promoting tumor growth, angiogenesis, and immunosuppression.
Immunological Transition from Antitumor Inflammation to Tumor-Promoting Immunosuppression in HCC
During the transition of HCC from precancerous lesions—such as liver cirrhosis, hepatic adenoma, and dysplastic nodules—to early-stage malignancy, macrophages, particularly liver-resident KCs, play a pivotal role in shaping the TME. Through alterations in their polarization states, origins, and functional reprogramming, these cells profoundly influence hepatic immune tolerance, inflammatory responses, and tumor progression.137-139
In the precancerous stage, chronic inflammation of the liver drives the persistent activation and M1 polarization of KCs. These pro-inflammatory macrophages secrete cytokines such as TNF-α and IL-1β, thereby sustaining inflammatory responses and suppressing aberrant cell growth.28,140 As the disease advances, the polarization profile of KCs may shift toward an M2 phenotype, characterized by immunosuppressive properties and the secretion of anti-inflammatory cytokines, including IL-10 and TGF-β, which promote hepatic immune tolerance and contribute to the establishment of an immunosuppressive microenvironment supportive of tumor initiation. 48
In early HCC, M2-polarized macrophages facilitate tumor cell proliferation, migration, and angiogenesis through the secretion of cytokines and growth factors, thereby supporting tumor growth and metastasis. 141 These immunosuppressive macrophages also impair the cytotoxic function of CD8+ T cells, promoting tumor immune evasion and enabling cancer cells to escape immune surveillance. 142
At the initial stages of hepatocarcinogenesis, the hepatic immune microenvironment still exhibits certain anti-tumor activity. Cytotoxic CD8+ T cells, NK cells, and predominantly M1-type macrophages remain active, suppressing tumor cell proliferation and dissemination through antigen presentation and the release of effector cytokines such as IFN-γ and IL-12, thereby maintaining immune surveillance.137,138 However, as tumor cells continue to proliferate and accumulate genetic and epigenetic abnormalities, HCC gradually acquires immune evasion capabilities. Tumor cells downregulate major histocompatibility complex (MHC) class I/II molecules, upregulate immune checkpoint ligands such as PD-L1, and reprogram glucose and lipid metabolism to weaken immune recognition and cytotoxic clearance.139,143
Concurrently, hepatic myeloid immune cells—particularly KCs and monocyte-derived macrophages (MoMFs)—undergo profound phenotypic and functional reprogramming driven by tumor-associated signals, including IL-10, TGF-β, and CSF-1.48,138 As the disease progresses, the macrophage population shifts from a predominantly M1 pro-inflammatory phenotype to an M2 immunosuppressive and pro-tumorigenic state. These M2 macrophages secrete high levels of anti-inflammatory cytokines (eg, IL-10, TGF-β), growth factors (eg, VEGF, PDGF), and MMPs, promoting tumor invasion, metastasis, ECM remodeling, and neovascularization, thereby providing both structural and signaling support for tumor expansion.59,144,145
Moreover, M2 macrophages further compromise host anti-tumor immunity by suppressing CD8+ T-cell cytotoxicity, disrupting dendritic cell antigen presentation, and recruiting regulatory T cells as well as myeloid-derived suppressor cells (MDSCs).146-148 These events collectively contribute to the establishment of a profoundly immunosuppressive microenvironment that facilitates tumor immune escape and progression.
Overall, the transition from an “inflammatory–antitumor” to an “immunosuppressive–protumor” microenvironment represents a pivotal immunological event in the early development of HCC. 143 The repolarization and functional reprogramming of macrophages not only attenuate immune surveillance but also facilitate angiogenesis and stromal remodeling, establishing the foundation for tumor immune escape, local invasion, and the formation of a pre-metastatic niche. This immunological shift underscores the dual role of macrophages in HCC progression and provides critical insight into potential targets for immunomodulatory and antitumor therapies.
Tumor Stemness in Hepatocellular Carcinoma
Tumor stemness refers to the characteristics of tumor cells that resemble normal stem cells, including self-renewal, differentiation potential, high tumorigenicity, and resistance to therapy. These cells, known as cancer stem cells (CSCs), play a critical role in tumor initiation, progression, metastasis, and treatment resistance. In HCC, CSCs and TAMs interact closely, and their crosstalk is pivotal in tumor development, progression, and metastasis formation.
The rapid proliferation of HCC cells is closely tied to the formation of the immune microenvironment. In the early stages of tumor progression, the immune microenvironment can exert anti-tumor effects. However, as tumor cells evade the immune system, an immunosuppressive microenvironment form. The interaction between TAMs and CSCs weakens anti-tumor responses, 149 enhances immune escape, and plays a central role in tumor progression. Fan et al, using single-cell and spatial analyses, revealed the co-localization of CSCs and SPP1-overexpressing macrophages in hypoxic regions. HCC patients with high CSC levels exhibit accumulation of SPP1+ macrophages (Macro_SPP1), which express metalloproteinases (MMP9, MMP12, and MMP7) regulated by Hypoxia-Inducible Factor 1 Alpha (HIF1-α). The presence of Macro_SPP1 and CSCs in hypoxic tumor regions is associated with poor prognosis in HCC. 150 S100 calcium-binding protein A9 (S100A9), an inflammation-related secreted protein, is significantly upregulated in TAMs and correlates with poor survival in HCC patients. 151 S100A9 enhances the stemness-like characteristics of tumor cells by activating the nuclear factor NF-κB signaling pathway via the receptor for advanced glycation end-products (RAGE). Treatment with S100A9 leads HCC cells to recruit more macrophages via CCL2, indicating a positive feedback loop between TAMs and HCC cells. TAMs upregulate S100A9 secretion, 151 enhancing the stemness-like properties of HCC cells and promoting tumor development and metastasis. Dysregulation of lncRNA H19 is associated with HCC. Forced overexpression of H19 attenuates miR-193b-mediated suppression of multiple oncogenic drivers (EGFR, KRAS, PTEN, and IGF1R) and the MAPK1 gene, triggering EMT and stemness transformation in HCC. TAM-induced lncRNA H19 promotes HCC invasiveness by activating the miR-193b/MAPK1 axis, mediating crosstalk between HCC and the immune microenvironment, and leading to adverse clinical outcomes. 152 Periostin (POSTN), a member of the fasciclin protein family secreted by CSCs, significantly promotes M2 TAM recruitment in intrahepatic cholangiocarcinoma (iCCA). 153 CSCs in iCCA release various molecules, such as IL-13, OA, and IL-34, which guide macrophage precursors to adopt an M2-like pro-tumor phenotype. 154
The interaction between TAMs and CSCs is bidirectional and can involve sequential crosstalk. Targeting TAMs to modulate CSCs or targeting CSCs to influence M2 TAMs represents an emerging research focus. In-depth studies of CSC biology and the mechanisms of CSC-TAM interactions at various stages of tumor growth could play a critical role in suppressing HCC progression. Researchers have found that macrophages can be engineered with chimeric antigen receptors (CARs) to target tumor antigens, improving the treatment of solid tumors.155,156 Therefore, exploring new therapeutic targets by leveraging CSC-TAM interaction mechanisms holds promise for future breakthroughs.
M2 TAMs Promote Cancer Cell Proliferation, Invasiveness, and Metastasis
M2 TAMs play a significant role in anti-tumor responses and immune escape. M2 TAMs secrete growth-promoting cytokines that directly stimulate cancer cell proliferation. Epidermal growth factor (EGF), 157 platelet-derived growth factor (PDGF), 158 and TGF-β 158 activate receptor signaling pathways (eg, EGFR, PDGFR) on cancer cells, promoting DNA synthesis and cell cycle progression, thereby accelerating proliferation. IL-6 activates the JAK/STAT3 pathway, 159 inducing the expression of anti-apoptotic proteins and cell cycle proteins, enhancing cancer cell survival and proliferation. Cytokines such as TNF-α, Intercellular Adhesion Molecule 1 (ICAM-1), and IL-6 regulate the tumor immune microenvironment by modulating cancer cell EMT. 160 Additionally, activated M2 macrophages secrete CCL22, enhancing tumor invasiveness and inducing EMT through Smad2/3 and Smad1/5/8 activation and Snail upregulation, 161 thereby increasing cancer cell migration. CCL2, secreted by M2 macrophages, is closely associated with tumor stemness and EMT via the TGF-β1 and Wnt/β-catenin signaling pathways. 162 Cathepsin B, modified in lysosomes and secreted extracellularly, directly degrades the extracellular matrix and promotes cancer cell migration. Increased glucose metabolism in TAMs enhances O-GlcNAcylation of lysosomal cathepsin B, promoting cancer metastasis and chemotherapy resistance. 163 Exosomes secreted by M2 TAMs carry miRNAs such as miR-21-5p and miR-155-5p, which, upon transfer to colorectal cancer cells, downregulate BRG1 expression, promoting cancer cell migration and invasion. 164 In iCCA animal models, tumor cells accelerate the differentiation of THP-1 cells (a human acute monocytic leukemia cell line) into M2 macrophages, which secrete IL-10 to promote HCC cell growth, invasion, and EMT. 165 Accumulation of ROS and paracrine TNF from KCs leads to mitochondrial dysfunction and oxidative stress, potentially causing precancerous lesions. Blocking the ROS/TNF/JNK axis may be an effective therapeutic strategy to mitigate iCCA tumor growth. 166
Enhanced communication between TAMs and tumor-associated cells also promotes cancer invasion and metastasis. During cancer progression, CAFs, M2 macrophages, and regulatory T cells (Tregs) form an immune barrier against CD8 CTL-mediated anti-tumor immune responses, leading to CTL dysfunction and exhaustion, 167 thereby facilitating tumor cell proliferation and migration. M2 TAMs secrete immunosuppressive factors such as TGF-β, affecting NK cell metabolism by reducing glycolysis and OXPHOS, thereby inhibiting NK cell effector functions and diminishing the immune system’s ability to kill cancer cells.168,169 Co-culture of TANs and macrophages results in higher expression levels of oncostatin M and interleukin-11 compared to individual cultures. Crosstalk between TANs and TAMs enhances intrahepatic cholangiocarcinoma (ICC) proliferation and invasiveness through STAT3 signaling. 113
M2 TAMs Stimulate HCC Angiogenesis
Due to rapid proliferation, malignant tumors, including HCC, often experience intratumoral ischemia and secrete pro-angiogenic factors to promote angiogenesis, typically resulting in a rich blood supply. In addition to tumor cells, immune cells in the immune microenvironment contribute to angiogenesis. Studies have shown a positive correlation between tumor microvascular density and macrophage counts, highlighting the critical role of TAMs in early HCC neovascularization. 170 TIE2, a receptor for angiopoietin, transmits pro-angiogenic signals and identifies a subset of monocytes/macrophages with pro-angiogenic activity. These tumor-associated monocytes/macrophages (TEMs) primarily aggregate around vascular regions in tumor tissues and participate in HCC angiogenesis.171,172
M2 TAMs directly induce endothelial cell proliferation and maturation by secreting pro-angiogenic factors such as VEGF 173 and PDGF, 174 forming new blood vessels. The balance between human macrophage metalloelastase (HME) and VEGF gene expression significantly affects tumor angiogenesis.175,176 For example, cytokines derived from iCCA cells induce macrophage differentiation into M2 TAMs, increasing vasculature and VEGF expression. 113
VEGF produced by tumor tissues not only promotes angiogenesis but also drives macrophage polarization toward the M2 phenotype. 177 In turn, M2 TAMs secrete VEGF, further promoting angiogenesis. Thus, macrophage phenotype is positively correlated with HCC clinical prognosis. Studies show that CD14 inflammatory macrophages, stimulated by hepatitis virus-infected hepatocytes, secrete large amounts of IL-23, accompanied by upregulation of IL-23 receptors and a strong macrophage-associated angiogenic response. 178 CCR2 TAMs are more abundant at the highly vascularized margins of HCC, and their absence reduces pathogenic angiogenesis. 179
The hypoxic TME induces M2-like functional transformation of TAMs through direct effects, metabolic influences, lactic acidosis, angiogenesis, and matrix remodeling, enabling TAMs to participate in immunosuppression, angiogenesis, and other tumor-supporting processes. 180 The Warburg effect in the TME promotes a hypoxic microenvironment, stabilizing HIF-1α and upregulating VEGF, synergistically enhancing angiogenesis. 181 TAMs increase HIF-1α and Sema4D expression in cancer cells, influencing tumor growth and progression. 182 In clinical models, macrophage accumulation is associated with resistance to anti-VEGF therapy, 183 potentially due to downregulation of VEGFR-1 and VEGFR-3 and upregulation of pro-angiogenic genes. Therefore, combining VEGF blockade with macrophage inhibitors can enhance anti-VEGF angiogenic responses, offering a strategy to intervene in HCC progression.
HCC Cells Regulate TAM Metabolic Reprogramming
In HCC, M2 TAMs undergo metabolic reprogramming regulated by HCC cells through various mechanisms, including glucose metabolism, lipid metabolism, and amino acid metabolism. These reprogramming events promote the pro-tumor phenotype of M2 TAMs, supporting HCC progression and metastasis.
Under normal conditions, glucose metabolism pathways include glycolysis, the TCA cycle, and the pentose phosphate pathway, with most normal cells relying on aerobic respiration, where the TCA cycle in mitochondria is the primary glucose metabolism pathway. M1 macrophages preferentially use aerobic glycolysis, exhibiting TCA cycle disruption and impaired OXPHOS, while M2 macrophages rely more on mitochondrial OXPHOS.12,184 Interestingly, tumor cells exhibit active glycolysis even under oxygen-sufficient conditions, consuming large amounts of glucose and producing lactate, a phenomenon known as the Warburg effect. M1 macrophages induced by LPS share similar Warburg metabolism with tumor cells, characterized by reduced OXPHOS and ATP levels, increased glucose consumption and lactate synthesis, and elevated expression of key glycolytic enzymes such as hexokinase and glucose-6-phosphate dehydrogenase. 185 Elevated glycolysis regulates PD-L1 levels and promotes M2 polarization.186,187 However, recent studies show that macrophage populations with Human Leukocyte Antigen – DR isotype high expression (HLA-DRhigh) and CD86high expression in HCC tumors exhibit a PD-L1 glycolytic phenotype, where intrinsic glycolytic metabolism confers anti-tumor properties to PD-L1 macrophages. 188 Tumor-derived soluble factors, such as hyaluronic acid fragments, regulate glycolysis in peritumoral monocytes by upregulating 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), a key glycolytic enzyme that coordinates cellular metabolism and induces PD-L1 expression, weakening CTL responses in tumor tissues. 189 Additionally, HCC cell culture supernatants activate the Wnt2b/β-catenin/c-Myc signaling pathway, enhancing glycolysis in M2 TAMs. 190 HCC cell-derived exosomal PKM2 induces metabolic reprogramming in monocytes, phosphorylating STAT3 and upregulating differentiation-related transcription factors. These findings suggest that PKM2 promotes monocyte differentiation into macrophages and reshapes the TME. 191 Regulating key enzymes or exosomes in the TME can modulate macrophage polarization, reducing tumor invasion and metastasis.
Lipid metabolism significantly influences macrophage polarization. Different fatty acids alter lipid synthesis and catabolism in hepatocytes by inducing changes in macrophage polarization phenotypes, affecting liver lipid metabolism balance. 192 Lipid content varies across different macrophage polarization states. After LPS-induced polarization to M1, lipid content differs significantly from normal cells, with differential lipids primarily associated with fatty acid synthesis, sphingolipid metabolism, glycerolipid metabolism, phospholipid synthesis, and inositol phospholipid metabolism pathways. 193 M2 macrophages and TAMs exhibit abnormal lipid accumulation, accompanied by activation of mitochondrial and peroxisomal fatty acid oxidation (FAO) pathways. 194 Lipid metabolism reprogramming in TAMs is essential for macrophage polarization and HCC development. Downregulation of receptor-interacting protein kinase 3 (RIPK3) in TAMs reduces ROS levels and promotes fatty acid metabolism via PPAR activation, facilitating M2 TAM accumulation and polarization. 195 Additionally, under stress, macrophages increase fatty acid transport to mitochondria, efficiently utilizing FAO pathways to produce ATP. 196 Targeting key points in lipid metabolism reprogramming offers potential strategies for inhibiting HCC tumor progression.
Specific amino acids are directly associated with different macrophage polarization states, and amino acid changes in tumor cells can affect TAMs. Glutamine, an important carbon and nitrogen source in M1 macrophages, is involved in metabolic reprogramming during macrophage activation. 197 Deficiency in hepatic glutaminase reduces the ability of macrophages to produce glutamate through glutamine catabolism, limiting TCA cycle substrate replenishment, decreasing glycolysis-related amino acids (eg, threonine), and increasing OXPHOS-related amino acids (eg, tyrosine, leucine), favoring the metabolic profile of M2 macrophages. 198 Lysyl oxidase-like 4 (LOXL4), an amine oxidase involved in ECM remodeling, is upregulated during hepatocarcinogenesis in mice fed a choline-deficient, L-amino acid-defined diet. 199 Tumor cell-derived LOXL4 may promote macrophage infiltration into the liver and accelerate tumor growth. Glutamine metabolism is critical for macrophage activation, with glutamine catabolism producing α-ketoglutarate (αKG) to support activation. 199 Solute carrier family 7 member 11 (SLC7A11) increases CSF1 expression via the αKG-HIF1α cascade, leading to intratumoral TAM and MDSC accumulation. 200 Tryptophan-derived microbial metabolites mediate anti-tumor immunity in TAMs and activate the aryl hydrocarbon receptor, suggesting a potential therapeutic strategy. 201 The specific mechanisms by which amino acid metabolism in macrophages influences polarization and tumor progression remain unclear and require further investigation.
HCC Cells Induce TAM Polarization by Modulating the Mechanical Environment
HCC cells can influence the mechanical environment, such as matrix stiffness and ECM remodeling, to promote the polarization of TAMs toward the pro-tumor M2 phenotype. In China, most HCC cases result from hepatitis B, progressing through the “hepatitis B–cirrhosis–HCC” trilogy, during which the liver tissue gradually hardens. HCC patients with severe cirrhosis typically have poorer prognosis and shorter median survival times. Matrix stiffness is used to assess the histopathological features of HCC. 202 The physical properties of the ECM (eg, stiffness) regulate TAM phenotypes by activating cell membrane mechanoreceptors and corresponding mechanotransducers. 203 Excessive collagen secretion and crosslinking increase matrix stiffness, enhancing macrophage mechanosensing. 204 In turn, M2 macrophages activate fibroblasts into myofibroblasts, which secrete large amounts of collagen to promote matrix deposition while remodeling the ECM by regulating the balance between MMPs and their inhibitors. 205 Thus, matrix stiffness and macrophages regulate each other in a feedforward manner, ultimately mediating tumor invasion and metastasis. CAFs and TAMs are key cells involved in forming metastatic niches, playing critical roles in ECM alterations that promote cancer cell adhesion and growth. 206 Using decellularized tissue models, studies have shown that the ECM can directly educate TAMs to adopt an immunomodulatory phenotype associated with tumor immunosuppression and poor prognosis. 207 This suggests that ECM remodeling may regulate tumor progression by influencing TAM polarization. Studies indicate that a 3D gel-like microenvironment induces monocyte adhesion and differentiation via MAPK-NF-κB activation. 208 Activation of the integrin β5-FAK-MEK1/2-ERK1/2 pathway promotes the expression of HIF-1α and LOXL2 in polarized macrophages mediated by matrix stiffness. 209 High expression of Nogo-B in TAMs of HCC patients is closely associated with yes-associated protein 1 (YAP)/tafazzin (TAZ)-mediated M2 polarization. 210 Therefore, exploring the impact of matrix stiffness on macrophage polarization in the tumor microenvironment represents a highly innovative perspective.
Macrophage Polarization and Clinical Prognostic Models in Hepatocellular Carcinoma
HCC is one of the most common lethal malignancies worldwide, 211 ranking as the fifth most prevalent cancer in men and the seventh in women. 212 It is typically diagnosed at advanced stages, leading to poor prognosis. 213 Despite global efforts to improve HCC clinical management, high postoperative recurrence and metastasis rates result in persistently unfavorable outcomes for affected patients. 214 Therefore, exploring the relationship between macrophage polarization and HCC, and developing clinical prognostic models based on macrophage polarization, holds significant importance for improving patient prognosis.
The PD-1/PD-L1 pathway negatively regulates T-cell activation and function, leading to suppression of immune responses in cancer patients. 215 PD-1 binding induces the formation of PD-1/TCR inhibitory microclusters, which recruit SHP2 molecules, dephosphorylating multiple members of the TCR signaling pathway and effectively inhibiting T-cell activation. The PD-1/PD-L1 pathway has also been implicated in promoting M2 polarization of quiescent macrophages in the TME. 216 IFNγ and/or 1TLR agonists induce PD-1 and PD-L1 expression in macrophages, and the interaction between PD-1 and PD-L1 promotes M2 polarization, particularly of M2a and M2c macrophages. 217 Guolong Xu et al investigated the combined administration of a Listeria-based HCC vaccine (Lmdd-MPFG) and anti-PD-1 immune checkpoint blockade antibodies. The Lmdd-MPFG vaccine activates the NF-κB pathway in TAMs via TLR2 and MyD88 pathways, recruiting p62 to activate autophagy, thereby reprogramming M2-polarized TAMs into M1-polarized TAMs. 218 Simultaneously, it shifts the cytokine profile in the TME toward an anti-tumor spectrum, enhancing T-cell responsiveness to PD-1 blockade. 218 The combination of Lmdd-MPFG and PD-1 blockade exerts synergistic anti-tumor effects by modifying TAMs in the TME and relieving T-cell inhibitory signals, offering a novel potential strategy for HCC treatment. RNA sequencing revealed elevated Gasdermin E (GSDME) levels in HCC patients resistant to anti-PD-1 therapy, with TAMs being the primary cells expressing GSDME. Studies demonstrated that the non-N-terminal fragment of GSDME in macrophages specifically interacts with PDPK1, affecting its phosphorylation and activating the PI3K-AKT pathway, promoting M2 macrophage polarization. 124 Given the regulatory role of TAMs in the TME, targeting TAMs can enhance tumor immunotherapy. 57 Reprogramming or repolarizing TAMs to exhibit anti-tumor properties, combined with anti-PD-1 antibodies, may improve immunotherapy outcomes.219,220 In HCC samples with normal or elevated serum CA19-9 levels, elevated CA19-9 levels correlate with increased M2 macrophage levels. Compared to IgG-treated tumors, anti-PD-1-treated tumors exhibit more M2-polarized macrophages and fewer M1-polarized macrophages, a phenomenon reversed by anti-CA19-9 administration. 221 Thus, CA19-9 may influence immunotherapy efficacy by affecting macrophage polarization in the TME, and targeting CA19-9 could improve immunotherapy outcomes.
HCC progression is associated with macrophage polarization states. Akira Asai et al, using an HCC chimera model (humanized chimeric mouse model), found that M2b TAMs predominate in advanced HCC patients, representing a critical target for HCC immunotherapy. 222 Genes related to macrophage polarization (MP) in HCC patients are also a research focus. Han Chen et al developed a prognostic model based on 4 MP-related genes (SCN4A, EBF3, ADGRB2, HOXD9, CLEC1B, and MSC) to calculate risk scores for each patient, effectively predicting HCC prognosis, assessing invasiveness, and guiding drug therapy. SCN4A was identified as a suppressor gene in HCC. 223
To improve early diagnosis and treatment of HCC, recent studies have identified several potential biomarkers. In HCC, bone morphogenetic protein and activin membrane-bound inhibitor (BAMBI) is associated with tumor proliferation, EMT markers, glycolysis, fatty acid biosynthesis and degradation pathways, and immune checkpoint regulation. 224 BAMBI promotes M1 macrophage polarization and is linked to the expression of key genes involved in glucose and lipid metabolism, while modulating genes in the TGF-β and Wnt signaling pathways, exerting multifaceted roles in HCC. 224 Thus, BAMBI may serve as a prognostic biomarker for HCC. In recent years, exosomes have emerged as critical players in HCC progression and as novel targets for diagnosis, prognosis, and therapy. Formimidoyltransferase-cyclodeaminase (FTCD), an exosome secreted during HCC progression, shows a significant positive correlation with macrophage infiltration. FTCD stimulates M1 macrophage polarization and inhibits HCC cell proliferation. 225 Therefore, FTCD is a potential exosome-related biomarker for HCC diagnosis, prognosis, and treatment.
Recent studies have advanced the understanding of macrophage polarization and its prognostic significance in HCC. Increasing evidence indicates that the polarization state of macrophages within the TME is highly plastic and critically influences HCC progression and immunotherapeutic response. However, current research faces several limitations. Most studies remain restricted to small, single-center cohorts without large-scale or multicenter validation, limiting model reproducibility and clinical generalizability. Existing prognostic models, primarily based on transcriptomic or single immune parameters, often lack multi-omics integration—such as proteomics, metabolomics, and ST—thereby failing to capture the dynamic and complex nature of the TME. Moreover, macrophage polarization is frequently oversimplified into the binary M1/M2 paradigm, neglecting its continuous spectrum and reversible transitions, which constrains the interpretation of immune response heterogeneity.
Future research should therefore pursue multicenter validation across diverse populations and integrate multi-omics approaches to elucidate the dynamic regulation of macrophage polarization and its crosstalk within the immune microenvironment. Therapeutic strategies targeting macrophage polarization—such as TAM reprogramming combined with PD-1 blockade—warrant further clinical evaluation, particularly in immunotherapy-resistant HCC. Additionally, artificial intelligence and machine learning may facilitate the development of robust, multidimensional risk prediction models for survival and treatment outcomes.
In summary, macrophage polarization research offers promising insights and targets for precision medicine in HCC, yet its clinical translation remains limited by insufficient mechanistic depth, data integration, and validation. Continued multidisciplinary efforts are essential to bridge the gap between experimental discovery and clinical application.
Drugs Targeting Macrophage Polarization
HCC, a leading cause of cancer-related deaths worldwide, faces challenges such as drug resistance and high recurrence rates. As key immune cells in the TME, macrophages directly influence tumor progression and immunotherapy responses through their polarization states. Numerous preclinical and clinical studies are exploring drug interventions to modulate macrophage polarization, particularly by inhibiting M2 polarization or promoting M1 polarization to enhance anti-tumor immune responses.
TAM Recruitment
Depleting TAMs is critical for impeding HCC progression.
137
Most macrophage-targeted therapeutic strategies focus on blocking the recruitment of the monocyte-macrophage system from the bloodstream to the TME or reprogramming TAMs into immunostimulatory M1 macrophages. The CCL2/CCR2 axis facilitates TAM recruitment to the TME, with KCs being a primary source of CCL2.226,227 CCL2 is a prognostic factor in HCC, and inhibiting the CCL2/CCR2 axis suppresses M2 polarization of TAMs, impairing tumor progression in mouse HCC models by inducing T-cell-mediated anti-cancer immune responses.227,228 Neutralizing CCL2 with antibodies (anti-mouse CCL2; clone C1142) reduces inflammatory myeloid cell levels, inhibits IL-6 and TNF-α expression, and suppresses tumor growth in HCC models
229
(Figure 4). BMS-813160, a CCL2/CCR5 inhibitor, is being tested in clinical trials for its ability to block the recruitment of TAMs and prevent their transformation into the immune-suppressive M2 type. By blocking the CCL2/CCR5 signaling pathway, BMS-813160 stops monocytes from entering the tumor and becoming M2 macrophages, which contribute to immune suppression. This effect is particularly promising when combined with immune checkpoint inhibitors like nivolumab, an anti-PD-1 antibody.
230
miR-26a reduces M-CSF levels via PI3K/Akt signaling, decreasing TAM recruitment to the TME.
231
Emactuzumab, a CSF1R inhibitor, is currently being investigated in Phase I clinical trials for its ability to inhibit the recruitment and survival of TAMs. Although not specifically targeted at HCC, it has shown promise in other cancers, including glioblastoma multiforme, where it blocks TAM infiltration and reprograms the tumor immune microenvironment.
232
GC33, a novel recombinant fully humanized monoclonal antibody targeting human glypican-3 (GPC3), inhibits M2-polarized macrophage recruitment to impair HCC progression and has been evaluated in phase I clinical studies233,234(Figure 4). GPC3 overexpression promotes tumor progression and immunosuppression by activating pathways such as Wnt/YAP/Hedgehog, while GC33 binding blocks these signals, reducing M2 polarization-related inputs and shifting the TME toward an inflammatory (M1-like) state.
235
Inhibition of TAM recruitment.
TAM Depletion
Combination studies focus on modulating TAMs in cancer immunotherapy.
236
Sorafenib,
237
an oral multi-kinase inhibitor approved for HCC treatment, impairs the ability of polarized macrophages to induce EMT and migration in HCC.
238
In addition to the HGF-MET axis, sorafenib also modulates the TME in HCC through multiple targets and signaling pathways,239-243 as shown in Figure 5, and has already been utilized in the clinical treatment of liver cancer. (Figure 5). Zoledronic acid (ZA) accelerates TAM apoptosis.
244
ZA-mediated TAM suppression enhances sorafenib’s efficacy in cancer treatment, angiogenesis inhibition, and HCC lung metastasis suppression.
245
Lenvatinib modulates cancer immunity in the TME by reducing TAMs and shows enhanced anti-tumor activity via the IFN signaling pathway when combined with PD-1 blockade.
246
Additionally, ZA reduces TAM infiltration and angiogenesis in HCC rat models.
247
Targeting macrophage recruitment by inhibiting chemokines such as CCL2 or growth factors like CSF1/CSF1 receptor248-250 requires combination strategies to achieve therapeutic efficacy. Modulating TAM function via TREM2 (triggering receptor expressed on myeloid cells 2) inhibition
251
or small-molecule inhibitors of phosphatidylinositol 3-kinase γ (PI3Kγ) also relies on combination with checkpoint inhibitors (CPIs).
252
CD47 antibody blocking CD47-SIRPα interaction enhances macrophage-mediated phagocytosis of HCC cells.253-255 Wu et al. demonstrated that GPC3-targeted chimeric antigen receptor (CAR) T-cell therapy combined with sorafenib enhances anti-tumor effects in immunocompetent and immunodeficient HCC mouse models at subpharmacological doses, partly by promoting IL-12 secretion in TAMs and cancer cell apoptosis.
256
The serine/arginine-rich splicing factor (SRSF) family, critical splicing factors and RNA-binding proteins, is essential for immune evasion and anti-PD-1 resistance via the SRSF10/MYB/glycolysis/lactate axis. Selective inhibitor 10C1, targeting SRSF10, may overcome anti-PD-1 resistance in HCC.
257
Specific xCT knockout reduces TAM recruitment and infiltration, inhibits M2 polarization, and enhances ferroptosis activity in TAMs, significantly increasing PD-L1 expression in macrophages and improving anti-PD-L1 therapy efficacy.
258
Leukotriene A4 hydrolase (LTA4H) reduces M2 TAM polarization and delays HCC progression by targeting the HNRNPA1/LTBP1/TGF-β axis. Combined targeting of TGF-β and PD-1 significantly improves immunotherapy resistance in LTA4H-knockout tumors.
259
Clinical trials indicate that blocking TGF-β or its receptors exerts potent anti-tumor effects across various cancers, including HCC, with acceptable safety profiles.
260
Recent advances involve combining anti-TGF-β antibodies or receptor inhibitors (eg, galunisertib) with immune checkpoint inhibitors (ICIs), triggering robust anti-tumor immunity and tumor regression in mouse models.
261
M7824, a bifunctional fusion protein targeting PD-L1 and TGF-β, activates innate and adaptive immune responses and induces tumor regression in mouse models.
262
JX-594, a targeted oncolytic poxvirus expressing granulocyte-macrophage colony-stimulating factor (GM-CSF), selectively replicates and destroys cancer cells. Jeong Heo et al. demonstrated that JX-594 followed by sorafenib exhibits anti-tumor activity in HCC, potentially sensitizing tumors to subsequent VEGF/VEGFR inhibitor therapy.
263
Sorafenib modulates TME and immune responses.
TAM Reprogramming
Reprogramming TAMs is also considered a strategy to impair HCC progression. Leveraging macrophage plasticity to “re-educate” pro-tumor macrophages into immunostimulatory phenotypes activates host anti-tumor immunity. Miro Viitala et al. found that macrophages lacking the scavenger receptor common lymphatic endothelial and vascular endothelial receptor-1 (Clever-1) become immunostimulatory, tilting the immunosuppressive TME toward inflammation and activating endogenous anti-tumor CD8+ T cells
264
(Figure 6). Immunotherapeutic blockade of Clever-1 achieved similar effects, comparable to PD-1 checkpoint inhibition.
264
Norcantharidin (NCTD) has been shown to inhibit HCC progression by promoting macrophage M1 polarization through the upregulation of miR-214, thereby enhancing anti-tumor immune responses.
265
In addition, GSK872 (RIPK3 inhibitor) or FAO inhibition effectively reversed the immunosuppressive phenotype of macrophages and suppressed HCC tumorigenesis by remodeling TAM function.
195
Baicalin has been shown to increase M1 macrophage polarization
266
and enhance pro-inflammatory cytokine production, directly inducing TAM and M2-like macrophage repolarization to M1, inhibiting HCC (Figure 6). 8-Bromo-7-methoxychrysin (BrMC), another factor that impairs CD163 (a marker of M2-polarized macrophages) expression,
267
reverses M2 polarization of TAMs in HCC by inhibiting NF-κB activation (Figure 6). PLX3397, a highly specific competitive inhibitor of CSF-1R tyrosine kinase, inhibits xenograft model growth by enhancing M1 macrophage polarization, while blocking CSF-1R reduces tumor growth
268
(Figure 6). TAM reprogramming.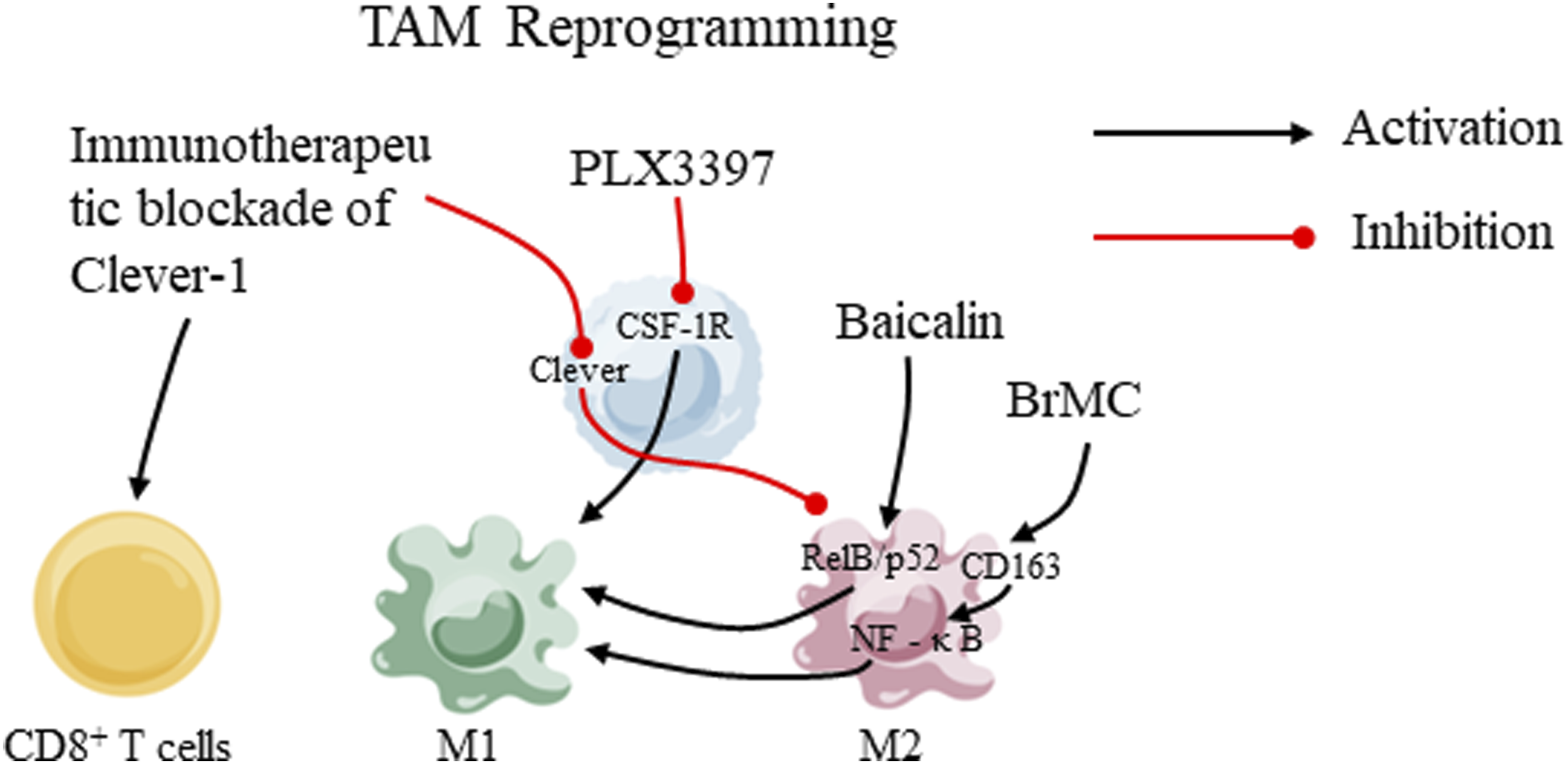
Exosomes
Exosome-based delivery systems hold promise for targeted delivery to specific cell populations, expanding the therapeutic index.
269
Exosomes are extracellular vesicles (30-200 nm) released by all cells,
270
which can be engineered to deliver various therapeutic payloads to target cells, emerging as an efficient delivery system.271,272 Reprogramming TAMs toward a pro-inflammatory M1 phenotype is a novel approach to induce anti-tumor immunity. The M2 phenotype is controlled by key transcription factors, such as STAT6. An engineered exosome therapeutic candidate, exoASO-STAT6, delivers antisense oligonucleotides (ASOs) targeting STAT6, selectively silencing STAT6 expression in TAMs.
273
Administration of exoASO-STAT6 induces nitric oxide synthase 2 (NOS2, an M1 macrophage marker), remodeling the TME and eliciting CD8 T-cell-mediated adaptive immune responses. ExoASO-STAT6 is a novel exosome-based therapeutic that selectively targets tumor macrophages, generating potent anti-tumor activity
273
(Figure 7). Additionally, engineered CD147-CAR macrophages enhance phagocytosis in cancer.
274
HES-TG100-115-CDM-PEG micelles, a novel delivery system loaded with sorafenib and with TG100-115, a PI3Kγ inhibitor, showed synergistic effects in the treatment of HCC. The system improves targeted drug delivery by enhancing the responsiveness of the TME, thereby inhibiting HCC tumor growth while further suppressing the immunosuppressive activity of TAM and enhancing the antitumor effect of CD8+ T cells.
275
In HCC, high levels of CircFUT8 are modulated by human methyltransferase-like 14 (METTL14). Exosomal miR-628-5p extracted from M1 macrophages, when transferred to HCC cells, inhibits METTL14 expression, suppressing HCC progression.
276
Chen Yichi et al developed a smart exosome-like nanodrug, MPDA/ICG-loaded exosome-like nanoparticles (MPDA/ICG@M1NVs), to enhance near-infrared (NIR)-based synergistic photoimmunotherapy for HCC. These exosome-like nanoparticles reduce immunosuppression in the TME by inducing M2-to-M1 repolarization of TAMs, enhancing anti-tumor immune responses triggered by immunogenic cell death (ICD), significantly improving anti-tumor efficacy
277
(Figure 7). Exosomes also serve as carriers, collaborating with polyethylene glycol-coated iron oxide nanoparticles loaded with chlorin e6 (PIONs@E6), to enhance M1 macrophage polarization, improving anti-tumor immune responses in HCC
278
(Figure 7). Low-intensity ultrasound (LIUS)-processed H-Exos loaded with hydrophobic curcumin (H-Exo-Cur) demonstrate significant anti-tumor effects in HCC,
279
offering a potentially safe and effective approach for the clinical application of exosome-based immunotherapy in HCC. Exosome-based targeted delivery and therapy.
Strategies for Macrophage Reprogramming in HCC
With the advancement of single-cell omics, ST, and immuno-microenvironment profiling technologies, the plasticity of TAMs in HCC has been recognized as a pivotal determinant of immune therapeutic response and tumor progression. Recent research has shifted from mechanistic exploration to multidimensional therapeutic modulation, establishing TAM reprogramming as a core strategy in reshaping the HCC immune microenvironment. These approaches encompass 5 major dimensions: source control, functional reprogramming, metabolic modulation, microenvironmental remodeling, and precision delivery, forming an integrated framework for macrophage-targeted immunomodulation.
At the source-control level, inhibition of the CCR2/CCL2 and CCR5 axes effectively prevents the recruitment and infiltration of peripheral monocytes into tumor sites, thereby attenuating the establishment of immunosuppressive TAM populations and creating a favorable foundation for immune activation. 48 Concurrently, blockade of the CSF1/CSF1R signaling pathway, which governs TAM survival and M2 polarization, not only reduces TAM density but also enhances the efficacy of ICIs and anti-angiogenic therapies, representing one of the most validated myeloid-targeting strategies across cancer types. 280
At the functional reprogramming level, interventions focus on both restoring phagocytic activity and promoting pro-inflammatory activation. The CD47–SIRPα axis, a canonical “don’t eat me” signal, when inhibited, reinstates macrophage-mediated phagocytosis and antigen presentation. In preclinical HCC models, blockade of this pathway has demonstrated potent synergism with ICI or chemotherapy. 281 Moreover, targeted inhibition of PI3Kγ, TREM2, and the CD39/CD73 adenosine pathway can reverse the immunosuppressive phenotype and promote M1 polarization, offering new avenues to overcome anti-PD-1 resistance.282-284 Activation of innate immune sensors such as TLR and STING further reprograms TAMs toward a pro-inflammatory phenotype, with particular promise in post-radiotherapy or TACE contexts. 48
In terms of metabolic modulation, interventions targeting lipid and purine metabolism represent emerging frontiers in TAM reprogramming. Suppression of FAO or cholesterol esterification shifts macrophages from a “tissue-repairing” to an “antigen-presenting” phenotype. 285 Likewise, targeting purine metabolism and adenosine production establishes a dual regulatory axis linking metabolism to immunity, thus enhancing the intensity and durability of anti-tumor responses. 284
Microenvironmental remodeling represents another indirect yet effective reprogramming strategy. The combination of anti-VEGF/VEGFR therapy with ICIs not only normalizes aberrant vasculature but also remodels myeloid cell dynamics, facilitating improved immune infiltration. 48 Furthermore, local treatments such as TACE or radiotherapy induce immunologic reprogramming within the TME, revealing a peri-interventional “immune activation window” that can be exploited by combining inhibitors targeting TREM2, PI3Kγ, or the adenosine pathway. 283
Finally, innovations in precision delivery and macrophage engineering have expanded the translational potential of TAM-targeted therapy. CD206-directed nanocarrier or exosome systems enable the targeted delivery of siRNA or innate immune agonists (eg, STING/TLR activators) specifically to M2-like TAMs, achieving efficient hepatic enrichment with minimal systemic toxicity. 286 Meanwhile, CAR-macrophage (CAR-M) technology represents a paradigm shift toward cellular immunoengineering; notably, GPC3-targeted CAR-M has shown robust anti-tumor efficacy in HCC models, highlighting the future potential of in vivo immune cell programming. 287
In summary, TAM reprogramming in HCC is transitioning from single-target interventions toward multimodal, synergistic, and personalized therapeutic paradigms. The integration of anti-angiogenic agents, ICIs, and myeloid-targeted strategies into an ecosystem-based, stratified therapeutic framework holds promise for achieving more durable and precise anti-tumor immune responses in clinical practice.
Drugs and Research Progress Related to Macrophage Polarization in HCC Treatment. Categorized Into Drugs Targeting Macrophages, Exosomes, and Clinically Relevant Drugs, With Descriptions of Their Targets and Mechanisms
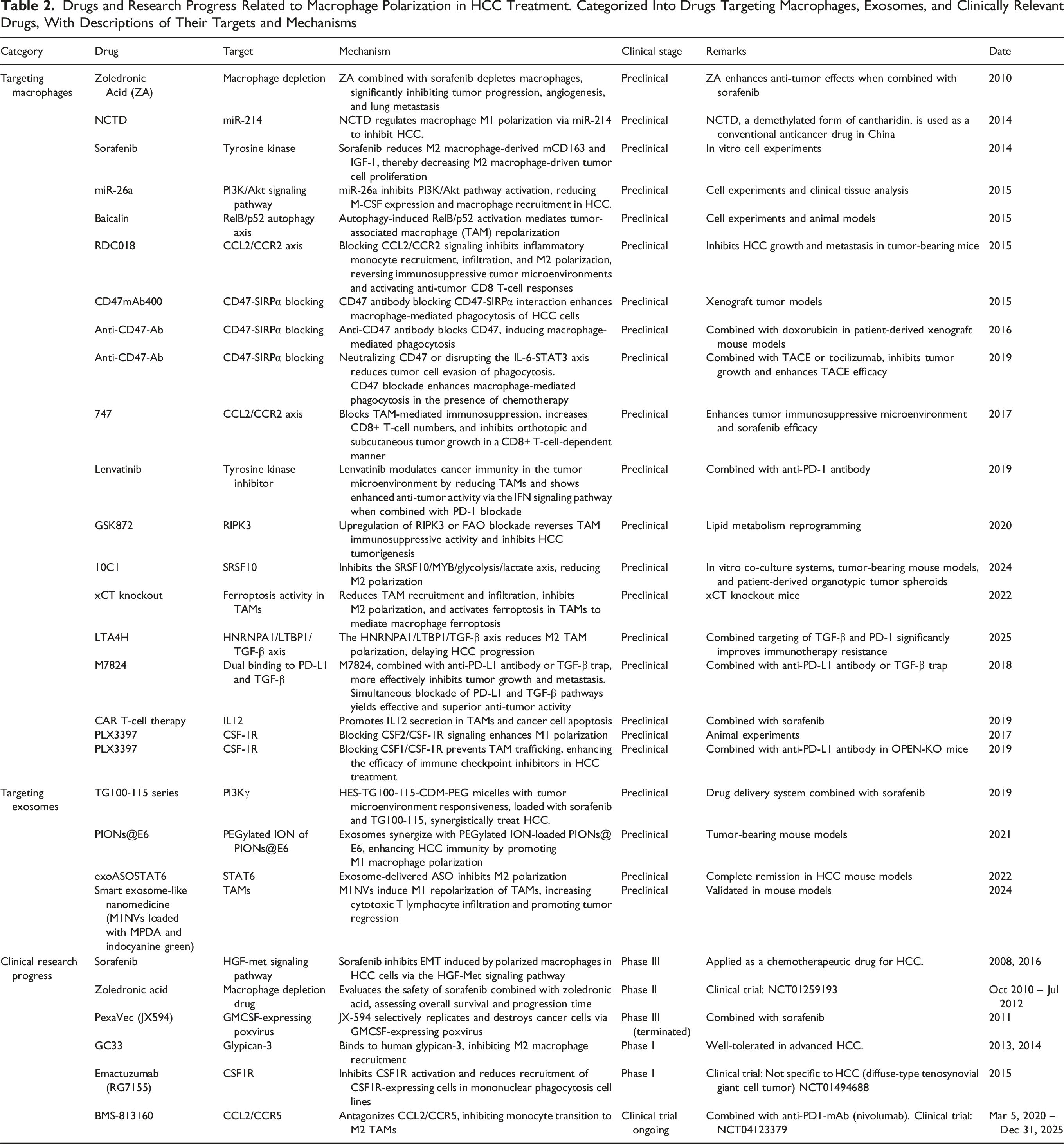
Future research should focus on integrating single-cell multi-omics, ST, and metabolic profiling to delineate the dynamic states and functional plasticity of TAMs under therapeutic pressure. Developing combination regimens that couple TAM reprogramming with PD-1/PD-L1 blockade, anti-VEGF/VEGFR therapy, or TACE-based immunomodulation may yield more durable and synergistic clinical outcomes. Furthermore, innovations in precision drug delivery and immune-cell engineering—such as CD206-targeted nanocarriers or GPC3-directed CAR macrophages—offer new directions for personalized macrophage-targeted therapy. Collectively, a deeper mechanistic understanding and rational integration of TAM-modulating strategies hold the key to transforming macrophage biology into clinically actionable immunotherapy for HCC.
Conclusion
Macrophage polarization represents a central regulatory axis in the pathogenesis of HCC, profoundly influencing tumor initiation, progression, angiogenesis, metabolic reprogramming, and therapeutic response. Liver-resident KCs and infiltrating monocyte-derived macrophages display remarkable phenotypic plasticity, transitioning along a continuum from pro-inflammatory, anti-tumor to immunosuppressive, pro-tumor states in response to cytokines, exosomes, metabolic cues, and biomechanical signals within the TME. Beyond the traditional M1/M2 paradigm, emerging single-cell and ST studies have revealed distinct TAM subpopulations—such as SPP1+, TREM2+, and MARCO+ macrophages—that construct hypoxia- and metabolism-dependent immunosuppressive niches, attenuate cytotoxic immune activity, and are strongly associated with poor prognosis and immunotherapy resistance.
Clinically, macrophage-centered prognostic models and therapeutic strategies—including blockade of macrophage recruitment (CCL2/CCR2, CSF1/CSF1R), depletion of immunosuppressive TAMs, and reprogramming toward M1-like phenotypes—offer promising avenues to enhance the efficacy of immune checkpoint inhibitors, anti-angiogenic agents, and locoregional treatments. Nonetheless, translation into clinical practice remains hindered by limited multicenter validation, incomplete integration of multi-omics data, and insufficient understanding of spatially and temporally dynamic macrophage states within the TME.
Future efforts should therefore emphasize large-scale, prospective, multicenter validation of macrophage-related prognostic models in diverse populations, coupled with standardized endpoints and analytical frameworks. Integrative multi-omics and spatial profiling—including single-cell transcriptomics, proteomics, metabolomics, lipidomics, and radiomics—will be essential to elucidate TAM lineage trajectories and intercellular communication networks during disease progression and therapeutic intervention. Mechanistic dissection of noncanonical TAM subsets (eg, SPP1+, TREM2+, MARCO+) should be pursued to identify druggable vulnerabilities while preserving beneficial myeloid functions. In parallel, mechanobiological regulation of macrophage states—through matrix stiffness, extracellular matrix remodeling, and YAP/TAZ signaling—warrants exploration using stromal-targeted agents such as LOXL inhibitors in combination with macrophage-reprogramming therapies.
Furthermore, insights into metabolic regulation—including glycolysis, fatty acid oxidation, cholesterol/purine metabolism, and lactate/adenosine signaling—should be leveraged to develop rational combination regimens that pair metabolic modulators (PI3Kγ, CD39/CD73, FAO inhibitors) with ICIs or anti-VEGF/VEGFR therapies. Innovative delivery and cell-engineering approaches, such as TAM-targeted nanocarriers or exosomes (eg, CD206-directed ASO/siRNA cargo) and GPC3-directed CAR macrophages, hold substantial potential but require optimization for pharmacokinetics, specificity, and manufacturability. Meanwhile, the application of artificial intelligence and machine learning to integrate clinical, pathological, and multi-omics datasets can facilitate real-time risk prediction and adaptive therapeutic decision-making.
By integrating these multidimensional approaches into unified translational frameworks—spanning molecular characterization, targeted modulation, and predictive analytics—the field is poised to transition from descriptive macrophage biology to precision macrophage medicine in HCC. This paradigm shift will not only deepen mechanistic understanding but also enable durable, clinically actionable immunotherapeutic strategies, ultimately improving patient outcomes in hepatocellular carcinoma.
Footnotes
Acknowledgments
We thank Mingquan Pang, Haijiu Wang and Zhixin Wang for their valuable contributions to the conceptualization, review, and editing of this manuscript. We also acknowledge the project support from Zhixin Wang and the financial support from Haijiu Wang. The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Ethical Considerations
This study does not involve human participants or animals, and therefore no ethical approval is required.
Author Contributions
Conceptualization, Xiaoqing Fu, Haijiu Wang and Zhixin Wang; Method-ology, Xiaoqing Fu; Investigation, Xiaoqing Fu; Writing original draft preparation, Xiaoqing Fu; Review and editing, Mingquan Pang, Haijiu Wang and Zhixin Wang; Supervision, Zhixin Wang and Mingquan Pang; Project administration, Haijiu Wang, Zhixin wang; Funding acquisition, Haijiu Wang. All authors have read and agreed to the published version of the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by Mechanism Research on Replication License Factor CDT1 Gene Poly-Morphism in Genomic Instability of Hepatocellular Carcinoma, grant number 82160466 and Correlation Between CDT1 Single Nucleotide Polymorphism Expression and Polarization Status of Tumor -Associated Macrophages in Hepatocellular Carcinoma, grant number CXCY202407.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.