
Abstract
Introduction
Neuroblastoma (NB) is one of the children’s most common solid tumors, accounting for approximately 8% of pediatric malignancies and 15% of childhood cancer deaths. Somatic mutations in several genes, such as ALK, have been associated with NB progression and can facilitate the discovery of novel therapeutic strategies. However, the differential expression of mutated and wild-type alleles on the transcriptome level is poorly studied.
Methods
This study analyzed 219 whole-exome sequencing datasets with somatic mutations detected by MuTect from paired normal and tumor samples.
Results
We prioritized mutations in 8 candidate genes (RIMS4, RUSC2, ALK, MYCN, PTPN11, ALOX12B, ZNF44, and CNGB1) as potential driver mutations. We further confirmed the presence of allele-specific expression of the somatic mutations in NB with integrated analysis of 127 RNA-seq samples (of which 85 also had DNA-seq data available), including MYCN, ALK, and PTPN11. The allele-specific expression of mutations suggests that the same somatic mutation may have different effects on the clinical outcomes of tumors.
Conclusion
Our study suggests 2 novel variants of ZNF44 as a novel candidate driver gene for NB.
Keywords
Introduction
With the development of precision medicine, discriminating genomic factors have gained prognostic and therapeutic implications. Many gene mutations, including somatic and germline mutations, are identified using whole-exome, genome, or transcriptome sequencing. Both have improved our understanding of carcinogenesis and influenced the development of cancer treatment plans, including neuroblastoma (NB). NB is one of the most common solid tumors in children, accounting for approximately 8% of all pediatric malignancies and 15% of childhood cancer deaths. 1 A four-center case-control study indicated that LIN28A SNPs (single nucleotide polymorphisms), especially rs34787247 G>A, may increase NB risk. 2 In contrast, a multi-center case-control study using 263 cases and 715 controls to examine the association of NRAS gene rs2273267 A>T polymorphism reduces NB risk in ethical Chinese children. 3 Nucleotide excision repair ERCC1/XPF genes surfaced in 4 polymorphisms for the genetic variations predisposed to NB risk upon screening Chinese children’s 393 cases and 812 controls. 4 Although these genomic studies have revealed specific features of the mutated genes in high-risk NB, their complicated molecular pathways remain unclear in allele-specific expression patterning of the protein-coding regions of genes. 5
Whole-exome sequencing (WXS, also known as WES) is a genomic technique that is gradually being optimized to identify mutations in increasing proportions of the protein-coding regions of genes. 6 It is now routinely used and has revealed some rare and common gene variants in NB. 7 The high-level amplification of MYCN on chromosome 2p24 was found in NB previously. 8 It occurs in approximately 20% of NB patients and indicates aggressive disease progress and a poor prognosis. Inhibitors that downregulate MYCN/MYC proteins can suppress NB tumor growth. 9 Mutations in the tyrosine kinase domain of anaplastic lymphoma kinase (ALK) have also been identified in NB. 10 A series of ALK tyrosine kinase inhibitors (TKIs) have been approved for use in ALK-driven cancers, including NB.11,12
ALK proteins can mediate different signaling outputs due to various properties, such as subcellular localization and protein stability. 13 Although most ALK-driven tumors respond dramatically to ALK-TKIs, most patients develop drug resistance. 14 However, the details of the resistance mechanism are not very clear. One thing for sure is that this phenomenon is not entirely due to the status of DNA mutation. That is to say, individual patients with the same somatic mutation can respond differently to targeted therapy. One of the potential reasons for variation in treatment response is the allele-specific expression of somatic mutations to model allele-specific expressions at the gene and SNP levels. Patients carrying the same mutations may express mutated alleles at different levels. Therefore, it is essential to investigate the expression levels of specific alleles at the mRNA level. This pattern is because proteins are made from mRNAs. Mutations in the DNA may result in very different allele expression patterns, ranging from no expression to dominance of one allele. Whole transcriptome sequencing (WTS, also known as RNA-sequencing or RNA-seq) is an emerging tool for profiling gene transcription and has received broad adoption in cancer genetics, with significant prognostic and therapeutic implications. 15 Therefore, we conducted this study to explore the allelic expression of somatic mutations in NB. The mutations of NB identified through DNA-seq, and RNA-seq were compared to confirm novel mutations in their allelic expression patterns, enabling us to propose new risk factors and potential therapeutic targets for NB.
Methods
Datasets
All of the sequencing data were obtained from the National Cancer Institute (NCI) Office of Cancer Genomics Therapeutically Applicable Research To Generate Effective Treatments (TARGET) NB project (https://ocg.cancer.gov/programs/target, assessed on 6 October 2020). The datasets were downloaded from The Cancer Genome Project (TCGA) Genomic Data Commons (GDC) Data Portal (https://docs.gdc.cancer.gov, assessed on 6 October 2020) using the GDC data transfer tool (https://gdc.cancer.gov/access-data/gdc-data-transfer-tool, assessed on 6 October 2020). A total of 127 RNA-seq and 219 WXS files were downloaded from the site. Of the downloaded sequences, both RNA-seq and WXS were available for 85 patients. In addition, 219 variant call format files (.vcf, specifically WXS.mutect2.raw_somatic_mutations.vcf) created using MuTect2 16 were downloaded for the patients on which WXS was performed to compare to our analysis of the raw sequencing files.
Variant Analysis for WXS Samples
Each VCF file from the WXS samples, ANNOVAR,
17
annotated variants (Figure 1A) was created. For each annotated VCF, we filtered the variants that were not exonic, synonymous SNVs, and those for which the maximum frequency in the population is > .001 in gnomAD.
18
We also filtered out the variants that had flags such as “alt_allele_in_normal,” “panel_of_normals,” or “germline_risk” in MuTect2
16
and required the remaining variants to have 'PASS' flags in more than one of the 219 WXS samples. Ultimately, using these criteria, we obtained 9 variants of the 8 genes. Workflow of the search for variants through filtering and analysis. The whole transcriptome sequencing (WTS): RNA-seq data. Exome sequencing (also known as whole-exome sequencing, WES, or WXS) is a technique for sequencing all the expressed genes in a genome. Population AF is the maximum allele frequency in the population obtained from GnomAD (the genome aggregation database). (A) Pipeline for filtering variants in WXS exome data from 219 patients. (B) Pipeline to call variants from 127 WTS RNA-seq datasets to analyze variants in the 8 genes for which variants meeting the criteria were found in (A). Shapes in dots: Inputs; boxes in dashes: outputs.
Variant Analysis for RNA-Seq Samples
As shown in Figure 1B, we wrote a python pipeline to call variants from 127 RNA-seq samples with BAM files as input [Binary Alignment Map (BAM) is the comprehensive raw data of genome sequencing; BAM is the compressed binary representation of SAM (Sequence Alignment Map), a compact and index-able representation of nucleotide sequence alignments]. We first used Picard 19 to mark duplicates and then used Genome Analysis Toolkit (GATK) 20 to split Reads with N in Cigar, generated and applied a recalibration table for Base Quality Score Recalibration (BQSR), and used HaplotypeCaller to call variants. After that, variants were filtered using GATK 20 with the options “-window 35 -cluster 3 -filterName Filter -filter ‘QD <2.0’ -filterName Filter -filter ‘FS >30.0.’” The filtered variants were annotated with ANNOVAR 17 for further analysis. Similarly, we filtered out non-exonic or synonymous SNV variants with >.001 maximum frequency in the population.
Additional methods included assessing the allele-specific or allele-imbalanced gene expression levels and manually examining the alignment files to locate candidates. We also obtained the GTeX data and the cBioPortal (The cBioPortal for Cancer Genomics website, https://www.cbioportal.org/, assessed on 6 October 2020) for comparative analyses.
Results
Clinical Characteristics
Clinical Characteristics in WXS- and WTS-Identified NB Samples.

Whole-Exome Sequencing Identifies Candidate Genes
A List of 9 Mutations Detected from MuTect2, Its Functional Impacts (To Protein), Population Frequency, Number of Occurrences in the Current Data Set, and the Respective Alt/Ref Reads Count.

RNA-Sequencing Analysis of Candidate Gene Allelic Expression
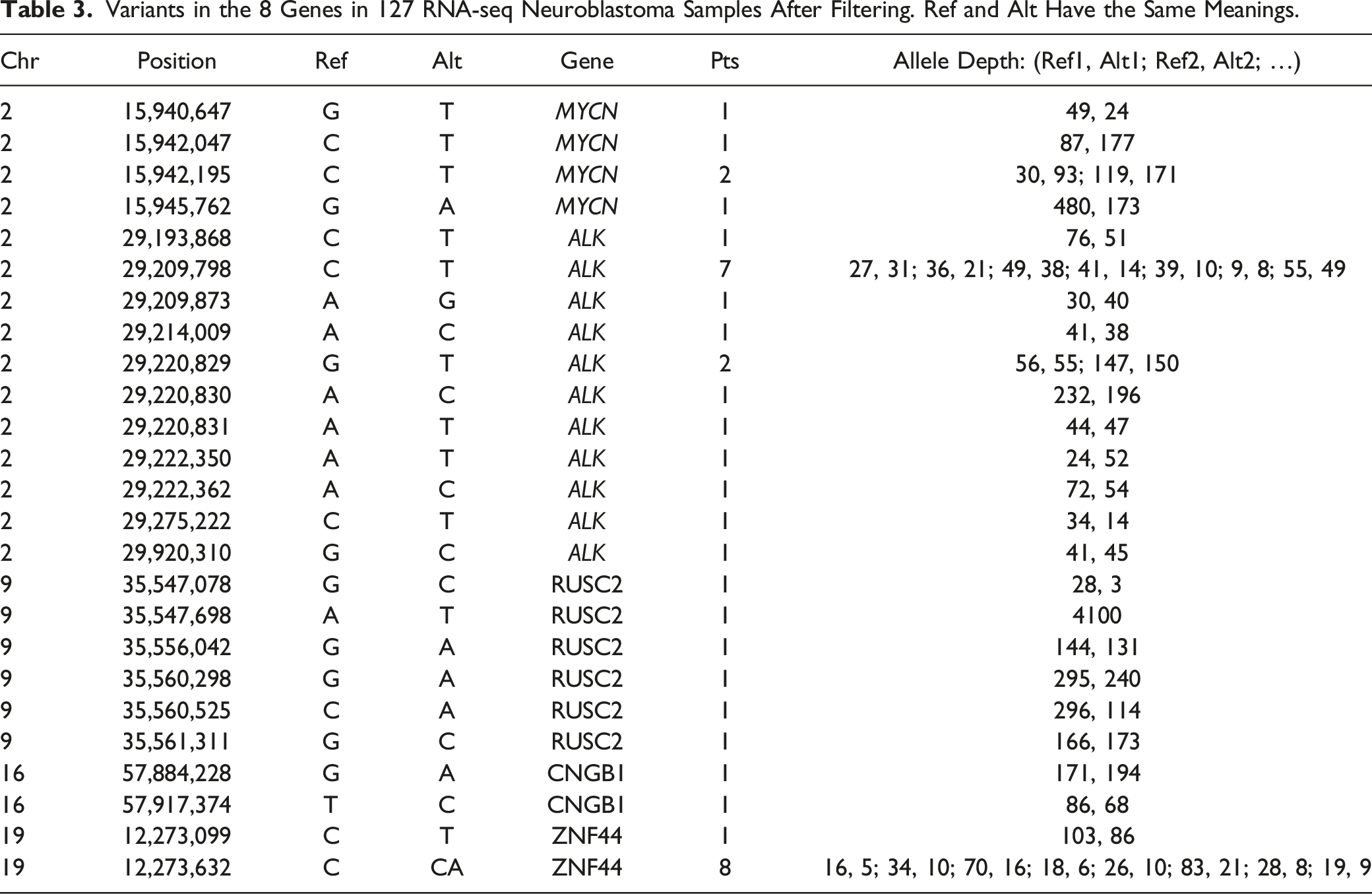
Variants in the 8 Genes in 127 RNA-seq Neuroblastoma Samples After Filtering. Ref and Alt Have the Same Meanings.

Patterns of 2 ALK variants in the ALK gene using IGV plot from RNA-seq data: (A) chr2:29209798/C>T with 3 out of 7 patient’s samples (the other 4 samples are shown in Supplementary Figure S4); (B): chr2:29220829/G>T with 2 patient’s samples.
Moreover, among these mutated sites, we observed 2 novel variants of ZNF44, which were not previously reported as associated with NB. Notably, the prevalence of the ZNF44 mutated allele at chr19: 12, 273, 632/C>CA (Figure 3) was much higher (8/127, 6.3%) than at other sites. After analyzing the clinical data, we found that the average tumor percentage in all samples was 80%, ranging from 60% to 90%. The average stroma ratio was approximately 20%, ranging from 10% to 40%, consistent with the previous report.
21
Thus, the change in ZNF44 expression was comparable and reliable across patients. As shown in Table 3, the allelic depths of 8 patients (normal vs tumor) were 16/5, 34/10, 70/16, 18/6, 26/10, 83/21, 28/8, and 19/9. The average fold change (normal vs tumor) was 3.0, ranging from 2.1 to 4.4. ZNF44 variant patterns by IGV plot for chr19:12,273,632/C>CA in the ZNF44 gene from 8 RNA-seq datasets. The box in black shows a region where insertions happen and which should result in the same insertion interpretation due to the polyA region.
Discussion
NB is a solid tumor that can develop from immature nerve cells in several areas of the body. It most commonly affects children and rarely occurs in adults. 1 This study analyzed WXS and RNA-seq data from NB patients to identify somatic mutations and their allele-specific expression. As most somatic mutations are identified from DNA-seq techniques such as WXS, the allelic expression of those mutations is often unknown. Proteins, the functional units of a live cell, are made from mRNA, so a somatic mutation may have very different effects on the cellular function that vary with its allelic expression profile. Our study confirmed multiple known NB mutations and identified ZNF44 (zinc finger protein 44) as a potentially actionable somatic mutation.
Our study explored 2 cohorts of NB patients with either WXS or WTS data available. The overlapping rates of the 2 cohorts were high: 38.8% (85/219) of the WXS group and 66.9% (85/127) of the WTS (Whole transcriptome sequencing) group. It has been reported that using WXS identification, mutation frequencies of somatic genes, including ALK (9.2% of cases), PTPN11 (2.9%), ATRX (2.5%), MYCN (1.7%), and NRAS (.83%) are significant in NB. 22 As expected, WXS revealed mutations in some of these genes, including ALK, MYCN, and PTPN11. The MYCN mutation rate (1.4%, 3/219) was similar to a previous report. However, the ALK (3.2%, 7/219) and PTPN11 (1%, 2/219) mutation rates were lower than previously reported. 23 Notably, in our study, RUSC2 presented the second-highest mutation frequency (3.7%, 8/219) in the analysis of WXS and was identified by WTS as well. RUSC2 is a protein-coding gene with mutations associated with mental retardation and microcephaly. CNGB1 and ZNF44 variants were revealed by both WXS and WTS as well. Although CNGB1 and ZNF44 are not commonly associated with NB, they both presented similar mutation frequencies (1%, 2/219) to MYCN in the WXS analyzed in this study. ZNF44 encodes a zinc finger protein, also known as a gonadotropin-inducible transcription factor (GIOT-2). ZNF44 is expressed in human organs and tissues at various levels (Supplementary Figure S1). Mutations in ZNF44 have been detected in cancers of the uterus, stomach, ovaries, melanoma, cervical, colorectal, lung, bladder, GBM (glioblastoma multiforme), liver, prostate, sarcoma, invasive breast carcinoma, head and neck, etc. (Supplementary Figure S2). However, mutations of this gene have not been reported in neuroendocrine tumors before; to the best of our knowledge, this is the first report on ZNF44 mutations found in neuroendocrine tumors (Supplementary Figure S3) and on 2 novel variants of ZNF44 associated with NB [ZNF44 mutated allele at chr19: 12, 273, 632/C>CA, Figure 3, Tables 2 and 3]. ZNF44 is reported to be involved in epilepsy susceptibility and binds a factor that is abundant in developing nervous tissue. 24 Thus, ZNF44 may play an as-yet-undetected role in neuroendocrine tumors such as NB. In our study, ZNF44 mutations were discovered in NB by both WXS and RNA-seq. Specifically, in the analyzed RNA-seq data, the prevalence of ZNF44 variant chr19: 12, 273, 632/C>CA, was 6.3%. Given that the samples’ average tumor and stroma percentages were highly consistent at 80% and 20%, respectively, the ZNF44 normal allele has expressed an average of 3.0-fold higher than the mutated allele, suggesting the ZNF44 variant might be a potential disease-related site.
As expected, there were some differences between WTS and WXS analysis. WXS identified a gene RIMS4 (Regulating Synaptic Membrane Exocytosis 4) with a high mutation frequency (8.7%, 19/219), but WTS did not detect this. More variants in known disease-related genes ALK (Supplementary Figure S4) and MYCN (Supplementary Figure S5) were detected in the WTS group. Still, far fewer were found by WXS, suggesting that WTS provided more information on gene variation. WXS and WTS may both be practical biotechnological methods with their advantages. WXS is considered the standard gold method and is routinely used in oncology. 7 However, it cannot reflect gene expression levels.
WTS has been hailed as a promising approach with distinct advantages, especially for determining transcriptome characteristics.
25
However, WTS is not suitable for the discovery of DNA mutations. Thus, combining WXS and WTS can provide complementary perspectives on gene mutations. In our study, an interesting finding from RNA-seq analysis was that in 1 patient sample harboring 2 different RUSC2 variants, the allelic expression levels of the normal vs mutated SNVs were quite different. Although the samples were from the same patient, the results were opposite, suggesting that the allelic expression levels of the 2 SNVs were not due to different ratios of normal and tumor cells in the sample but due to allele-specific expression. Proteins play bio-functional roles via both biological structure and expression levels. Therefore, both the mutation locations and their allelic expression levels may be related to the response to targeted therapy. However, the limitations of our study include the small sample size, limited clinical information, lack of original raw FASTQ files (any variant) to confirm indel alignment errors, and lack of original samples for clinical validation. A further, well-designed study with a more significant number of samples and clinical details is planned for the future (Figure 4). Proposed workflow of pediatric cancer for lifetime management. Collective information is based on the publications26-29 related to the single-cell subclonal evolution of pediatric tumors. This workflow might track 2 new variants of ZNF44 and validate them as a novel candidate driver gene for neuroblastoma as details in the discussions.
Highly relevant to our study, an Italian group aimed to determine the differential genetic landscapes between short survival (SS) and long survival (LS) in high-risk (HR) neuroblastoma (NB) (HR-NB) patients at stage M. 5 The significant percentage of patients who demonstrate rapid disease progression despite multimodal treatment presents one of the biggest problems for oncologists treating high-risk (HR-NB) patients. About 60% of these HR-NBs develop fatal conditions within 5 years of diagnosis. They focused on a cohort of stage M NB patients from the Italian NB Registry with complete clinical data, and follow-up over 10 years was considered, including SS (n = 14) and LS (n = 15). They found ZNF44 mutations in only 2 SS patients (#1965, #2578), about 14%, but not in LS patients. The percentage of mutated ZNF44 in the total of patients at the M stage, including SS and LS, is about 6%. They pointed out, “In SS patients, 4 genes (SMO, SMARCA4, ZNF44, and CHD2), all known to be expressed in neural tissues, were recurrently mutated and group-specific and carried particularly deleterious variants.” Intriguingly, 5 genes (CHD2, DIDO1, KRTAP4–8, ZNF44, and ZNF91) with SS-specific recurrence in the Italian cohort were mutated with SS-specificity also in Pugh cohort patients.
Nevertheless, ZNF44 had been barely investigated in previous research, even though the identification of gain-of-function mutations in the ALK receptor tyrosine kinase gene (also in our gene list) as the most common cause of familial NB led to the identification of identical somatic mutations and the rapid development of ALK as a tractable therapeutic target. 30 Similarly, we proposed a workflow of pediatric cancer for lifetime management with collective information based on the publications26-29 related to the single-cell subclonal evolution of pediatric tumors (Figure 4). This workflow might track 2 new variants of ZNF44 and validate them as a novel candidate driver gene for NB. However, the potential transcriptional control of downstream cascades by ZNF44 must be elucidated by either over-expression or knock-down of ZNF44 transcripts in vitro and in vivo. We can explore the DNA binding region or protein partners of ZNF44 by CHIPseq or pull-down assays. The potential loss-of-function of ZNF44 mutation could be illustrated by the 3D protein structure analysis using the alpha-fold-2 platform.
In summary, to date, few studies have explored gene mutations in NB, like simultaneously using both WXS and WTS. Our study revealed that these 2 methods present different perspectives and meaningful results. Specifically, we found that allele-specific expression assessed by RNA-seq can be quite different even for the same gene mutations, which underscores the importance of WTS in cancer research. Furthermore, we identified gene mutations through both methods, validating some well-known NB genes such as MYCN and ALK and discovering novel candidate genes such as ZNF44 variants. Importantly, mutations identified in this study in genes such as ZNF44 may provide new opportunities for diagnosis- and treatment-driven subclonal evolution, 27 which is worthy of further investigation (Figure 4). The candidate driver gene ZNF44 remains to be validated in the clinic.
Conclusion
We discovered 2 novel ZNF44 variants as novel candidate genes in NB.
Supplemental Material
Supplemental Material - RNA-Sequencing Combined With Genome-Wide Allele-Specific Expression Patterning Identifies ZNF44 Variants as a Potential New Driver Gene for Pediatric Neuroblastoma
Supplemental Material for RNA-Sequencing Combined With Genome-Wide Allele-Specific Expression Patterning Identifies ZNF44 Variants as a Potential New Driver Gene for Pediatric Neuroblastoma by Lan Sun, Xiaoqing Li, Lingli Tu, Andres Stucky, Chuan Huang, Xuelian Chen, Jin Cai, and Shengwen C. Li in Cancer Control.
Footnotes
Acknowledgments
We thank Jiang F. Zhong for his guidance and critical reading of the manuscript.
Author Contributions
Writing—initial draft preparation, XL and LS. Writing—review and revision, SCL; Conceptualization, JC, SCL; Methodology and formal analysis, LT and CH; Software and data curation, AS and XC. All authors have read and agreed to the published version of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study is partly supported by the Natural Science Foundation of Chongqing, China (cstc2020jcyj-msxmX1063). This work was also supported in part by the CHOC Children’s–UC Irvine Child Health Research Awards #16004004, CHOC-UCI Child Health Research Grant #16004003, and CHOC CSO Grant #16986004.
Ethical Approval
The data sets came from a public database without ethical issues. Specifically, all of the sequencing data were obtained from the National Cancer Institute (NCI) Office of Cancer Genomics Therapeutically Applicable Research To Generate Effective Treatments (TARGET) neuroblastoma project (https://ocg.cancer.gov/programs/target, assessed on 6 October 2020). The datasets were downloaded from The Cancer Genome Project (TCGA) Genomic Data Commons (GDC) Data Portal (https://docs.gdc.cancer.gov, assessed on 6 October 2020) using the GDC data transfer tool (![]() , assessed on 6 October 2020).
, assessed on 6 October 2020).
Data Availability
The results published here are based on data generated by the Therapeutically Applicable Research to Generate Effective Treatments (https://ocg.cancer.gov/programs/target, assessed on 6 October 2020) initiative phs000467. The data used for this analysis are available at ![]() .
.
Supplemental Material
The results published here are based upon data generated by the Therapeutically Applicable Research to Generate Effective Treatments (https://ocg.cancer.gov/programs/target) initiative, phs000467. The data used for this analysis are available at ![]() . Supplemental material for this article is available online.
. Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.