
Abstract
Fucosidosis is a rare lysosomal storage disorder characterized by deficiency of α-L-fucosidase with an autosomal recessive mode of inheritance. Here, we describe a 4-year-old Chinese boy with signs and symptoms of fucosidosis but his parents were phenotypically normal. Whole exome sequencing (WES) identified a novel homozygous single nucleotide deletion (c.82delG) in the exon 1 of the FUCA1 gene. This mutation will lead to a frameshift which will result in the formation of a truncated FUCA1 protein (p.Val28Cysfs*105) of 132 amino acids approximately one-third the size of the wild type FUCA1 protein (466 amino acids). Both parents were carrying the mutation in a heterozygous state. This study expands the mutational spectrum of the FUCA1 gene associated with fucosidosis and emphasises the benefits of WES for accurate and timely clinical diagnosis of this rare disease.
Introduction
Fucosidosis is a rare type of lysosomal storage disorder with an autosomal recessive mode of inheritance. 1 The disease is caused by germline mutations of the FUCA1 gene which encodes the 466 amino acid protein, α-L-fucosidase.1,2 The FUCA1 gene is located on the short arm (p) of chromosome 1 and consists of eight exons. 3 Homozygous or compound heterozygous mutations of FUCA1 gene result in deficiency of α-L-fucosidase which in turn leads to incomplete breakdown of fucose-containing glycoproteins, glycolipids and oligosaccharides.3–5 As a consequence, there is gradual and progressive accumulation of these fucose-containing compounds throughout the body.1,3
Fucosidosis presents with extensive phenotypic variability and affects the normal development of the skeleton, central nervous system (CNS), brain, skin and heart. 6 Interestingly, patients with fucosidosis have been reported to show abnormalities of the white matter with a reduced signal of the globus pallidus. 7 There are two major types of fucosidosis characterised by age and disease spectrum.6,8 Type I fucosidosis develops soon after birth and is severe and life-threatening; symptoms may include rapid psychomotor regression, high sweat salt, dysostosis multiplex, and severe neurological deterioration. 8 Patients with type I fucosidosis usually die within the first decade of life. 1 Type II fucosidosis is comparatively milder than type I and affected patients tend to have milder psychomotor retardation, normal sweat salinity and angiokeratoma. 6 Patients with type II fucosidosis usually have a longer life span than those with type I. 1
To date, approximately 120 cases of FUCA1 gene associated fucosidosis has been reported worldwide. 6 Although rare, most cases have tended to occur in Italy and Cuba and in the Hispanic-American population of New Mexico and Colorado. 6 We report here a case of fucosidosis in a 4-year-old Chinese boy.
Case report
Patient
A 4-year-old Chinese boy, born to non-consanguineous parents (Figure 1) had an uneventful perinatal history. However, at 12 months of age was admitted to our hospital with mild developmental regression, seizures and a reduced blood glucose level. By 18 months, he had become anorexic, was losing his ability to communicate verbally and had restricted body movement. At the age of 30 months, severe developmental regression and psychomotor retardation were observed.
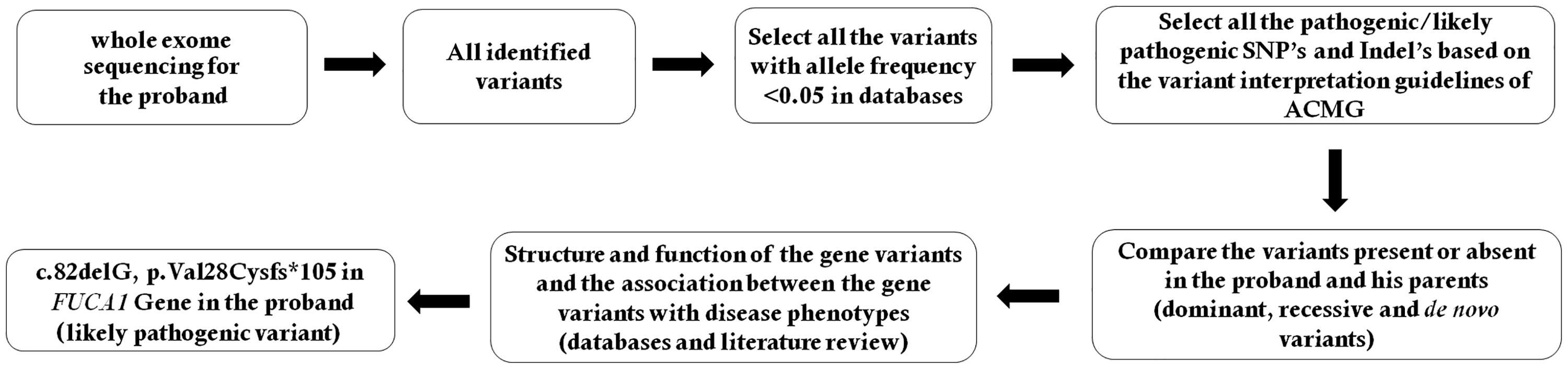
Schematic presentation of the filtering process for pathogenic mutations in all variants obtained by whole exome sequencing. SNP, single nucleotide polymorphism; Indel, small insertion or deletion; ACMG, American College of Medical Genetics and Genomics.
Physical examination of the proband at the age of three years showed abnormal skeletal development and a palpable liver. The patient had spastic quadriplegia and dystonia, and multiple joint contractures and loss of psychomotor responses were also observed. Although radiological imaging showed no abnormalities in the brain parenchyma, a brain magnetic resonance imaging (MRI) scan showed cerebral atrophy with abnormal T1 and T2 signal intensity over the globus pallidus. Normal cellular maturation was detected in a bone marrow biopsy. Abdominal ultrasonography showed slightly enlarged liver but urinalysis showed no mucopolysaccharides or organic acids. His blood counts, liver and thyroid function were normal. While electroencephalography identified seizures, no abnormalities were detected in nerve conduction velocity (NCV). A reduced activity of α-fucosidase in peripheral blood leukocytes (2.2 nmol/h/mg protein; normal control 24-162 nmol/h/mg protein) was observed but other lysosomal enzymes (i.e., α-glucosidase, α- and β-galactosidase and N-acetyl β-glucosaminidase) were within normal ranges. A liver biopsy was performed and degenerated, congested and mildly infiltrated hepatocytes were identified containing ceramide tetra- and penta-hexosides that had formed transparent swollen balloon-like (pseudo-gargoyle cells) structures.
Whole exome sequencing (WES) and Sanger sequencing of the proband was performed at the age of four years. DNA samples from the proband’s parents were also subjected to WES and Sanger sequencing. Genomic DNA was extracted from blood samples using a QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. This case report was approved by the Ethics Committee of Xi'an No.4 Hospital, Xi'an Obstetrics and Gynaecology Hospital, Xi'an, China. Signed patient consent was obtained from the patient’s parents before publication of this article.
Whole exome sequencing
Sequences were captured by Agilent SureSelect version 5 (Agilent Technologies, Santa Clara, CA, United States) according to the manufacturer’s protocols. This was followed by preparation of an exome enrichment library using Illumina HighSeq 4000 exome capture kit. The sequencing reads were aligned to GRCh37.p10 using Burrows-Wheeler Aligner software (version 0.59). Local realignment and base quality recalibration of the Burrows– Wheeler aligned reads were performed using the GATK IndelRealigner and the GATK Base Recalibrator, respectively. Single-nucleotide variants (SNV) and small insertions or deletions (indel) were identified by the GATK HaplotypeCaller. Variants were annotated using the Consensus Coding Sequences Database (20130630) at the National Centre for Biotechnology Information (NCBI).
Illumina pipeline was used for image analysis and base calling. Indexed primers were used for the data fidelity surveillance. Clean sequencing reads were aligned to the human reference genome (hg19) by the SOAP aligner (soap2.21) software. SOAPsnp (v1.05) software was used to assemble the consensus sequence and call genotypes in target regions
We selected variations obtained from exome sequencing with minor allele frequencies (<0.05) in any of the flowing databases: database of single nucleotide polymorphism (dbSNP), Hapmap, 1000 Genomes Project and our in-house database of 30,000 Chinese Han samples. Variant interpretation was performed according to American College of Medical Genetics and Genomics (ACMG) guidelines. 9 We also compared the remaining deleterious variations in the patient with variations carried by his unaffected parents and the gene’s functions with references in Online Mendelian Inheritance in Man (OMIM) catalogue and published literature. The filtering process, for the pathogenic mutations is shown in Figure 1.
Sanger sequencing
To validate putative mutation identified by WES, Sanger sequencing was performed. Primers were designed according to reference genomic sequences of the Human Genome from GenBank in NCBI. Primers were then synthesized and amplified. ABI PRISM 3730 automated sequencer (Applied Biosystems, Foster City, CA, United States) was used to sequence the Polymerase chain reaction (PCR) products. Sequence data comparisons and analysis were performed by DNASTAR SeqMan (DNASTAR, Madison, WI, United States).
The homozygous novel mutations identified through WES were validated by Sanger sequencing using the following primers: F1 5′-
Identification of a novel mutation in FUCA1 Gene
Both sequencing methods identified a homozygous novel single nucleotide deletion (c.82delG) in exon 1 of FUCA1 gene (Figure 2). This mutation will lead to a frameshift which will result in the formation of a truncated (p.Val28Cysfs*105) FUCA1 protein of 132 amino acids approximately one-third the size of the wild type FUCA1 protein (466 amino acids). Therefore, this is a ‘loss-of-function’ mutation.

Partial DNA sequences in the FUCA1 by Sanger sequencing of the patient (proband) and his parents. Arrows point to the mutation. The child (proband) had c.82delG, p.Val28Cysfs*105 mutation.
Sanger sequencing confirmed that this mutation was heterozygous in both parents (Figure 3). The mutation was not found in the Human Gene Mutation database (HGMD) or the ClinVar archive or in 100 normal healthy control individuals. Moreover, the homozygous novel mutation was not found in Exome Aggregation Consortium (ExAC), dbSNP, 1000 Genome Database or our in-house database of 30,000 Chinese Han samples.

Family pedigree. The black-filled square symbol (II) indicates the boy patient (arrow) and the half-filled symbols (I) show the unaffected carrier healthy parents. The novel mutation in the FUCA1 gene (c.82delG) was carried by both parents. WT, wild type.
Discussion
To our knowledge, there have only been five other reports of fucosidosis in the Chinese population. 10 Therefore, because fucosidosis is rare and associated with a wide spectrum of clinical and radiological findings, genetic screening would be the most effective method for timely and accurate clinical diagnosis when the disease is suspected. 6 In this report we describe a 4-year-old Chinese boy from non-consanguineous parents who presented with signs and symptoms of severe fucosidosis. Genetic testing using WES and Sanger sequencing identified a novel pathogenic mutation in his FUCA1 gene. The patient had a homozygous single nucleotide deletion (c.82delG) in the exon 1 of the FUCA1 gene. This mutation will lead to a frameshift which will result in the formation of a truncated FUCA1 protein (p.Val28Cysfs*105) of 132 amino acids which is approximately one-third the size of the wild type FUCA1 protein which consists of 466 amino acids. There are three conserved domains in the α-fucosidase enzyme; α-L-fucos (31-413 amino acids); AfuC (63–383 amino acids); Fucosidase-C (382–463 amino acids). The identified loss-of-function mutation in this report (c.82delG, p.Val28Cysfs*105) is thought to cause partial loss of the α-L-fucos and AfuC domains, and complete loss of the Fucosidase-C domain.
To-date, 36 disease-causing mutations have been reported in the FUCA1 gene. 11 Among them, most are missense/nonsense substitutions with deletions being less common.3,4,11,12 Therefore, our current findings expand the mutational spectrum of the FUCA1 gene associated with fucosidosis. Patients with fucosidosis either carry the homozygous or compound heterozygous mutations in FUCA1 inherited from their parents. This present case is unusual in that the family was non-consanguineous but the patient was carrying a homozygous mutation in FUCA1. Sanger sequencing confirmed that the mutation was present in both parents in a heterozygous state.
Although fucosidosis is rare, affecting only 1 in 200,000 births depending on the country, it shows significant clinical variability and its atypical presentation may lead to delays in diagnosis of this life-threatening disease. 6 Therefore, genetic screening may be a useful tool for identifying candidate genes and any disease-causing mutations., Indeed, genetic screening for fucosidosis has been helped by the availability of gene testing tools such as WES. In addition, we recommended carrier screening for parents of patients with fucosidosis to understand if the mutation is inherited or de novo.
In conclusion, our study expands the mutational spectrum of the FUCA1 gene associated with fucosidosis and emphasises the benefits of WES for the accurate and timely clinical diagnosis of this rare disease.
Footnotes
Acknowledgements
We are grateful to all families for their contributions. We thank the members of the Molecular Genetic laboratory for their assistance.
Declaration of conflicting interests
The authors declare that there are no conflicts of interest.
Funding
This study is supported by Science and Technology Program of Shaanxi Province, China (2017SF-062) and Xi'an Science and Technology Program, Shaanxi, China (2017117SF/YX011(9)).