
Abstract
Pituitary pars intermedia dysfunction (PPID) is a neurodegenerative disease of senior horses. Loss of dopaminergic inhibition of the melanotropes of the pars intermedia leads to increased concentrations of pro-opiomelanocortin (POMC)–derived peptides. Diagnosis is challenging due to pre-analytical variables, such as sample storage, handling, and time to analysis. Our objective was to develop an ELISA for ACTH measurement, which could ultimately form the basis for a stall-side equine ACTH test. We selected 2 ACTH-specific monoclonal antibodies, CBL57 and EPR20361-248, based on the recognition of separate epitopes, strong and rapid color change, and minimal background interference, including no cross-reactivity with themselves, each other, and the test reagents. CBL57 was chosen as the detection antibody (or secondary antibody). EPR20361-248, functionalized on superparamagnetic iron oxide beads, was chosen as the capture antibody (or primary antibody) to bind ACTH in plasma. The incorporation of magnetic beads marks the initial stage in establishing a platform that could potentially be utilized in the field, similar to other stall-side tests. The concentrations of antibodies, magnetic beads, and incubation durations were optimized. Our immunoassay detected unglycosylated rat recombinant ACTH. Further studies are ongoing to optimize and validate our assay using equine plasma and serum samples.
Pituitary pars intermedia dysfunction (PPID), also formerly known as equine Cushing disease, is the most common endocrinopathy affecting aged equids. Once thought to be an uncommon disease, PPID may affect >25–30% of the equine population >14-y-old.5,7
PPID-affected individuals share similar pathophysiologic characteristics with human Parkinson disease.2,6 In both diseases, there is progressive dysfunction of the hypothalamus-pituitary system, particularly in the dopamine-producing neurons. These neurons degenerate over time, leading to a reduction of dopamine production and release. Dopamine normally inhibits the production and release of pro-opiomelanocortin (POMC)–derived peptides from specific neurons, known as melanotropes, of the pars intermedia. Similar to humans with Parkinson disease, decreased dopamine levels in PPID-affected horses result in a loss of inhibitory control. Loss of inhibition leads to the overgrowth of melanotropes, which can manifest with or without the development of microadenomas or macroadenomas, and to an overproduction of POMC-derived peptides such as ACTH, alpha-melanocyte stimulating hormone, and corticotropin-like intermediate peptide (CLIP).2,5–7
Greater awareness of PPID has prompted both horse owners and veterinarians to conduct testing for PPID more frequently. The American College of Veterinary Internal Medicine has not yet published a consensus statement on the diagnosis and management of PPID in horses; however, a 2015 review does provide summarized findings and recommendations from many articles. 7 Also, detailed guidelines on the diagnosis and management of PPID in horses come from the equine endocrinology group at Tufts University, which are updated every 2 y. 1 The algorithm from 2023 indicates that measuring baseline ACTH is the preferred testing method in horses with severe and/or advanced clinical signs of PPID. ACTH testing requires that samples be sent to referral endocrinology laboratories, which increases the cost to owners and veterinarians and the time to diagnosis. Additionally, pre-analytical variables such as sample storage conditions, sample handling (e.g., time from blood sample collection to harvesting of serum), and time to analysis can significantly impact measured results.3,4,8
A rapid, sensitive, cost-effective, point-of-care test for equine ACTH measurement that can be performed stall-side, immediately after blood collection, would improve results, turnaround time, and has the potential to lower testing costs. Our hypothesis was that an ELISA can be developed for stall-side use. We had 2 main objectives: 1) to develop an ELISA for equine ACTH, and 2) to conduct preliminary validation of our ELISA by investigating the limit of detection (LOD) and linearity.
To ensure analytical sensitivity, we developed a sandwich ELISA using an anti-ACTH capture antibody and a biotinylated anti-ACTH detection antibody combination. Equine ACTH is not available commercially. Because ACTH is a highly conserved hormone among all species, we used recombinant, unglycosylated rat ACTH as the ELISA development target (ACTH [1-39], rat, 1 mg; Thermo Scientific). UniProt (https://www.uniprot.org/) was used to determine the homology between equine and rat ACTH. ACTH is a peptide hormone that is highly conserved among all species, and there are only 2 differences between equine and rat ACTH: rat valine 26 versus horse glycine 26, and rat asparagine 29 versus horse aspartic acid 29. 9
Basic assay construction was to sandwich the target ACTH between a detection and a capture antibody. The capture antibody binds ACTH from solution (e.g., blood, plasma, or serum), whereas the biotinylated detection antibody produces a colorimetric signal in the presence of streptavidin poly-horseradish peroxidase (SA poly-HRP; Fisher Scientific). Antibody selection was based on specificity of ACTH epitopes, minimal background signal, and low cross-reactivity between antibodies.
First, we used a direct ELISA to select a detection antibody. Two commercially available anti-ACTH antibodies with known N-terminus epitopes were chosen for evaluation: mouse monoclonal antibody (mAb) CBL57 (clone 57; MilliporeSigma) and mouse mAb AH26 (Fisher Scientific). Experiment 1 steps (performed in duplicate, total experiment time 24 h) were as follows:
Candidate antibodies were treated with an antibody clean-up kit (cat. 44600; ThermoFisher) according to the manufacturer’s instructions.
Purified antibodies were biotinylated (Lightning link biotin conjugation kit, cat. Ab201795; Abcam) according to the manufacturer’s instructions. Dilutions (0.5–2.0 µg/mL) of each antibody were prepared.
Wash buffer was prepared using 1 × PBS supplemented with 0.05% (v/v) Tween-20 (PBST).
Blocking buffer was prepared with 10% Superblock and PBST.
Two-fold serial dilutions (1.25–5 µg/mL) of recombinant rat ACTH, including a negative control with no ACTH added, were prepared in blocking buffer. Microplates (96-well, Polysorp, cat 473245; Nunc) were coated with each ACTH dilution (50 µL/well) and incubated overnight.
After a series of 3 washes with wash buffer, wells were blocked with blocking buffer for 1 h.
Blocking buffer was removed, and plates were incubated for 1 h with one of the biotinylated antibody preparations (50 µL/well), followed by a series of 3 washes using PBST.
Plates were then incubated for 1 h with SA poly-HRP, followed by a series of 4 washes using PBST.
Tetramethylbenzidine (TMB; Thermo Scientific) was added, and plates were transferred to a microplate reader (Model 680; Bio-Rad).
Optical densities (ODs) were recorded in a spreadsheet (Excel 16; Microsoft), and the antibody with the greater OD was chosen.
We chose biotinylated CBL57 as the detection antibody for all remaining experiments. For the second experiment, 3 proposed capture antibodies were evaluated using a sandwich ELISA: mouse mAb CBL56 (clone 56; MilliporeSigma), rabbit mAb EPR20361-248 (Abcam), and rabbit mAb EPR20361-235 (Abcam). Experiment 2 steps (performed in duplicate, total experiment time 24 h) were as follows:
Two-fold serial dilutions (1.25–5 µg/mL) of candidate no-capture antibodies were prepared in blocking buffer (10% Superblock and PBST). Each dilution was used to coat 96-well microplates (Polysorp; Nunc), which were incubated overnight, followed by a series of 3 washes using PBST.
Blocking buffer was added to each well, and plates were incubated for 1 h.
Blocking buffer was removed, and 1 µg/mL of rat recombinant ACTH was added to each well, incubated for 1 h, and then followed by a series of 3 washes.
Next, 1 µg/mL of biotinylated CBL57 was added to each well, incubated for 1 h, and followed by a series of 3 washes.
Plates were then incubated for 1 h with SA poly-HRP, followed by a series of 4 washes using PBST.
TMB was added, and plates were transferred to the microplate reader (Bio-Rad).
ODs were recorded in an Excel spreadsheet, and the antibody with the greatest OD was chosen.
On the basis of OD data, the capture antibody EPR20361-248 was deemed acceptable. Our next goal was to determine if the detection antibody (CBL57) and EPR20361-248 would exhibit cross-reactivity between them. To this end, we used 1 µg/mL of EPR20361-248, 1 µg/mL of CBL57, and decreasing concentrations (1 μg/mL; 100, 10, 1, 0 ng/mL) of the rat ACTH. This sandwich ELISA was performed in duplicate and run similarly to experiment 2 above (Fig. 1).
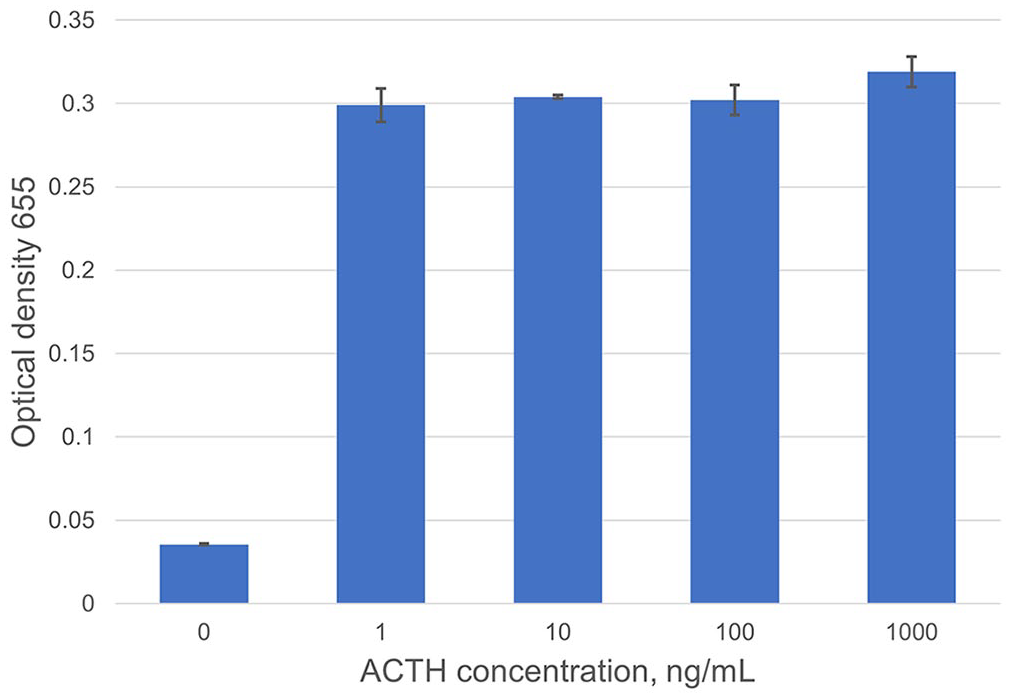
Investigation of detection and capture antibody cross-reactivity. Experiment without magnetic beads using 1 mg/mL of each antibody and varied ACTH concentrations. When no ACTH is present, there is minimal signaling, suggesting that antibodies do not cross-react with each other. Error bars denote SD between duplicates at each ACTH concentration.
Next, we incorporated magnetic beads (Pierce NHS-Activated; Thermo Scientific) to increase assay analytical sensitivity. Magnetic beads significantly reduce nonspecific binding of solutes and particles found in complex matrices such as plasma or serum, the ultimate intended sample type of our assay. Additionally, the spherical shape of beads provides a large surface area, enabling many molecules of capture antibody (EPR20361-248) to coat the bead and allowing small concentrations of ACTH within a sample to be captured. A series of washes removes unwanted sample matrix components and ensures that a pure solution of ACTH remains for detection. Magnetic beads also have the potential to facilitate testing in the field, as they eliminate the requirement for a centrifuge or bulky equipment.
Magnetic beads were coated with the capture antibody EPR20361-248 based on the manufacturer’s instructions. The ELISA was performed as follows, unless otherwise specified: washes were performed between steps, with the exception that no washing was performed between blocking incubation period and the addition of the ACTH. Volumes of 1 µL of magnetic beads were washed 3 times with PBST, then blocked for 1 h with 10% Superblock in PBST. The remaining steps were identical to experiment 2, steps 3–7. All experiments were run in duplicate.
Magnetic bead volume was varied to determine the lowest bead volume needed to reduce background signal, while still detecting physiologic concentrations of ACTH. Once the optimal magnetic bead volume was determined, a preliminary investigation of analytical sensitivity was conducted. We used 0.5 μL of antibody EPR20361-248–coated magnetic beads and followed steps 1 and 2 of experiment 2. ACTH concentrations of 1 and 10 pg/mL in blocking buffer were each added to 2 tubes with a negative control using only blocking buffer in one tube. The remaining steps were identical to experiment 2, steps 4–7 (Fig. 2).
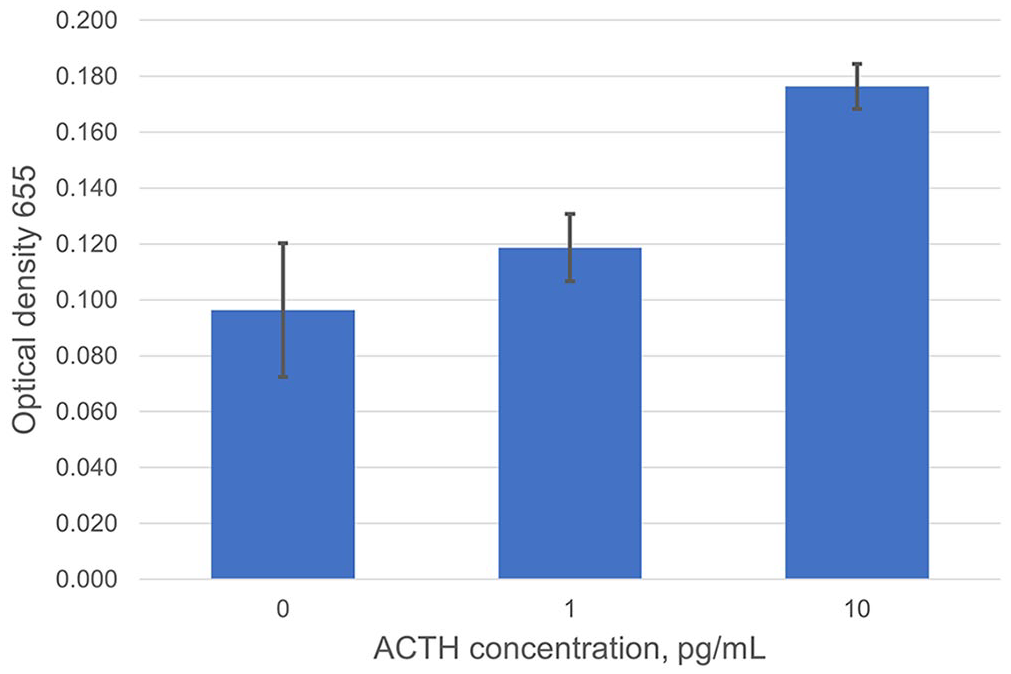
Investigation of analytical sensitivity. Preliminary data suggest the assay’s limit of detection is 1–10 pg/mL. Error bars denote SD between duplicates at each ACTH concentration.
Lastly, we investigated the linearity of the ELISA; 0.5 µL of capture antibody EPR20361-248–coated magnetic beads were used for the experiment and placed into 14 microcentrifuge tubes, washed 3 times, and then blocked with blocking buffer for 30 min. After incubation, supernatants were removed, and 500 µL of unglycosylated rat recombinant ACTH were added (10, 20, 40, 80, 100 pg/mL; 2 tubes at each concentration). The 0-pg/mL tubes contained 500 µL of blocking buffer, whereas blank tubes contained only magnetic beads and distilled water. The remaining steps were identical to experiment 2, steps 4–7 (Fig. 3).
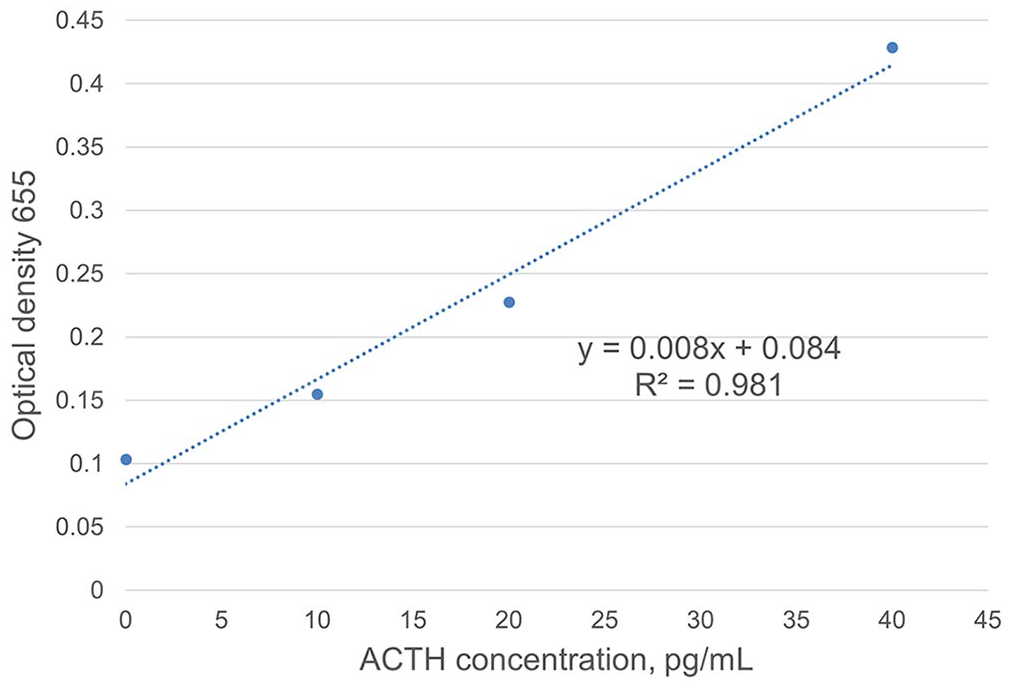
Investigation of linearity. Annotated blue dots are the averages of duplicates at each ACTH concentration. The trendline (dashed line) was determined using Excel spreadsheet trendline function and the corresponding equation of the line and R 2 value. Linearity of assay is established at 0–40 pg/mL.
Detection antibody CBL57 and capture antibody EPR20361-248 detected unglycosylated rat recombinant ACTH with minimal background interference, nonspecific binding, and minimal to no cross-reactivity with each other (Fig. 1). Optimal bead volume was determined to be 0.5 μL. Optimal incubation times were determined to be 30 min for all steps, except when ACTH was added and required 1-h incubation, which reduced time from 8 h to 6 h.
Using the detection antibody CBL57 and capture antibody EPR20361-248, we varied ACTH concentrations to evaluate the ability of the antibody pair to detect ACTH with minimal background signaling. There was no cross-reactivity between antibodies (Fig. 1). Analytical sensitivity is expected to be 1–10 pg/mL (Fig. 2); LOD experiments are needed to further define analytical sensitivity.
Linearity results were consistent, with a correlation (R 2 ) of 0.981, which was consistent through all runs (Fig. 3).
Our sandwich ELISA detects unglycosylated rat recombinant ACTH between 0 and 40 pg/mL in a linear manner; the interpretation threshold for baseline ACTH for equine PPID is expected to be ~15–30 pg/mL (depending on month or season), which is significantly <40 pg/mL. Assay optimization reduced run time from 8 h to 6 h and reduced the volume of beads needed to 0.5 µL. A 6-h run time is still too long for a stall-side, point-of-care test, and further refinement of assay conditions is needed.
Continued development of our ELISA as a stall-side equine ACTH assay requires the following, all of which are planned future experiments: investigation of whether the chosen detection and capture antibodies can detect ACTH in equine plasma (vs. the unglycosylated recombinant rat ACTH used here), confirmation of LOD, documentation of assay precision, investigation of prozone effect, and investigation of assay specificity (e.g., glycosylated vs. unglycosylated ACTH, ACTH vs. other POMC peptides). A major limitation of our project is that we did not evaluate equine samples for accuracy compared to the current Immulite 2000xpi or other reference methods (e.g., liquid chromatography–mass spectrometry).
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Partial funding for our project was provided by the ASVCP Share the Future Research Grant and by the Center of Excellence (COE) in Livestock Diseases and Human Health Research Fund, University of Tennessee.