
Abstract
Antimicrobial resistance (AMR) in pathogens important to aquatic animal health is of increasing concern but vastly understudied. Antimicrobial therapy is used to both treat and prevent bacterial disease in fish and is critical for a viable aquaculture industry and for maintenance of wild fish populations. Unfortunately, phenotypic antimicrobial susceptibility testing is technically difficult for bacteria recovered from aquatic animal hosts resulting in challenges in resistance monitoring using traditional methods. Whole-genome sequencing provides an appealing methodology for investigation of putative resistance. As part of the ongoing efforts of the FDA CVM Vet-LIRN to monitor AMR, source laboratories cultured and preliminarily identified pathogenic bacteria isolated from various fish species collected in 2019 from across the United States. Sixty-one bacterial isolates were evaluated using whole-genome sequencing. We present here the assembled draft genomes, AMR genes, predicted resistance phenotypes, and virulence factors of the 61 isolates and discuss concurrence of the identifications made by source laboratories using matrix-assisted laser desorption/time-of-flight mass spectrometry.
Aquatic animals are critical to the wellbeing of global society, particularly as a food source for humans and animals. As global aquaculture continues to grow to meet the demands of consumption, so too does the use of antimicrobials.11,19 In 2017, aquatic food animals accounted for nearly 5.7% (10,259 tons) of all antimicrobial usage among humans, and terrestrial and aquatic food animals globally. 19 Additionally, antimicrobial usage in aquaculture is expected to increase to ~13,644 tons by 2030. 19 Therefore, understanding the scope of antimicrobial resistance (AMR) in aquatic animal pathogens is vital for both animal and human health. A 2022 review of literature regarding AMR in aquaculture revealed a paucity of data, with only 32 studies performed in the United States between 1996 and 2021 despite a robust aquaculture industry. 4
Whole-genome sequencing (WGS) for the identification of AMR genotypes in veterinary diagnostic laboratories is supported by the U.S. Food and Drug Administration (FDA) Center for Veterinary Medicine (CVM) Veterinary Laboratory Investigation and Response Network (Vet-LIRN) in answer to the President’s Combating Antibiotic Resistant Bacteria (CARB) initiative in 2014. 5 Although limitations of inferring phenotypic antimicrobial susceptibility by genotyping still exist with WGS, this method supports the identification of AMR between veterinary laboratories and complements traditional characterization of AMR by antimicrobial susceptibility testing (AST) methods, which are more difficult for many aquatic bacteria. 8
We characterized genetic AMR and stress resistance genes to infer resistance phenotypes in 61 bacterial isolates from various fish species collected in 2019 from across the United States. In addition, we identified putative virulence factors. We also discuss here discordance of species-level identifications using matrix-assisted laser desorption/time-of-flight mass spectrometry (MALDI-TOF MS) and traditional methods with WGS.
Tissue from various fish species were submitted to 5 source laboratories (Cornell University–Animal Health Diagnostic Center [Ithaca, NY, USA], North Carolina State University–Veterinary Hospital Microbiology and Molecular Diagnostics Laboratory [Raleigh, NC, USA], Washington State University–Washington Animal Disease Diagnostic Laboratory [Pullman, WA, USA], Florida Department of Agriculture and Consumer Services–Bronson Animal Disease Diagnostic Laboratory [Kissimmee, FL, USA], and Mississippi State University–Aquatic Research and Diagnostic Laboratory [Mississippi State, MS, USA]) for preliminary identification, including salmonids (Oncorhynchus spp., n = 26), catfish (Ictalurus spp., n = 16), and other freshwater fishes (n = 12), as well as ornamental species: 2 black saddled tobies (syn. sharpnose puffer; Canthigaster valentini), 1 archerfish (Toxotes sp.), 1 piranha (species unknown), and 1 Koi fish (syn. common carp; Cyprinus carpio).
Bacterial cultures were derived from brain and/or kidney tissue homogenates and occasionally from skin and other mixed-tissue pools of diseased animals. Identifications were based on MALDI-TOF MS and/or other traditional biochemical methods by source laboratories, and bacterial isolates were either pelleted (following the procedures below) or frozen in glycerol and stored at −80 ± 10°C in preparation for transport to the sequencing laboratory. Frozen bacteria were subcultured to 5% sheep blood (Beckton Dickenson) and incubated at 29 ± 2°C in ambient air for 18–24 h. One colony was inoculated into trypticase soy broth in a 5-mL tube (Beckton Dickenson), and incubated under the same conditions. If an isolate did not grow adequately at 29°C, the incubation temperature for both the plated and broth growth was adjusted based on an appropriate temperature for that organism, if known; if unknown, the isolate was grown at both room temperature and at 35 ± 2°C in CO2. Following incubation, the broth was vortexed and two 1-mL aliquots were removed to sterile microcentrifuge tubes. The tubes were centrifuged for 2 min at 15,871 × g, the supernatant removed, and the resulting pellets frozen at −80 ± 10°C.
DNA was extracted using an automated magnetic bead–based process (MagMAX CORE; Thermo Fisher) and quantified with fluorometry (Qubit 2.0; Thermo Fisher). Genomic libraries were prepared and barcoded (DNA prep kit with Nextera CD indexes; Illumina), then sequenced (MiSeq platform, Reagent kit v3; Illumina) with 2 × 250 bp chemistry. Raw read sequences were uploaded to the NCBI SRA database (https://www.ncbi.nlm.nih.gov/sra), after meeting QC standards determined by GenomeTrakr network. 26 QC standards include R1 and R2 average read quality Q score ≥ 30, coverage ≥ 20-fold, and contig count ≤ 300.
Genomes were assembled by NCBI from the SRA repository using SKESA for de novo assembly and SAUTE for guided assembly using the AMRFinderPlus (https://www.ncbi.nlm.nih.gov/pathogens/antimicrobial-resistance/AMRFinder/) catalog of AMR genes (ARGs) as reference sequences to ensure sensitive and comprehensive cataloging of ARGs in each isolate.10,15,16,21,22 Assembled genomes were annotated by NCBI using the Prokaryotic Genome Annotation Pipeline (PGAP, https://www.ncbi.nlm.nih.gov/genome/annotation_prok/) and ARGs; stress tolerance genes and virulence factors were identified using AMRFinderPlus in “combined” mode via nucleotide, protein, and HMM search. For species-level identification, the assembled genomes were clustered by multi-locus sequence typing against a database of genomes curated by NCBI; isolates were placed into these organism clusters if they bore ≥ 25 matching alleles. However, this scheme was only applied to isolates with ≥ 1,000 conspecific organisms in the database; isolates with < 1,000 related organisms in the database were classified first by k-mer distance and then by single nucleotide variation clustering. These data were obtained from the NCBI Pathogen Detection Isolates Browser (https://www.ncbi.nlm.nih.gov/pathogens/isolates/) most recently on 2023 Mar 15 to ensure that the latest information was utilized (Suppl. Table 1). During curation, 2 samples (SAMN17105786, SAMN17106096) were excluded from publication in the Isolates Browser as a result of having too few loci covered by the assembly performed at NCBI. Two further samples (SAMN17136821, SAMN17106099) were still undergoing curation by NCBI staff. Although the species-typing was not validated by NCBI’s Pathogen Detection pipeline, results for the 4 isolates above were included as “unofficial” results (Suppl. Table 2).
Nine of the 57 curated isolates had published assemb-lies in GenBank (GCA_016414945.1, GCA_016414965.1, GCA_016414985.1, GCA_016611385.1, GCA_016611405.1, GCA_018038145.1, GCA_020765715.1, GCA_028991515.1, GCA_028991555.1) for which AMRFinderPlus results of these assemblies were published into the NCBI Pathogen Detection Microbial Browser for Identification of Genetic and Genomic Elements (MicroBIGG-E, https://www.ncbi.nlm.nih.gov/pathogens/microbigge/). The MicroBIGG-E dashboard contained further details for individual BioSamples, including the alignment statistics of assembled contigs and reference accession numbers for elements identified by the AMRFinderPlus pipeline.
For all isolates regardless of status at NCBI, read collections were downloaded from the SRA in bulk and imported into GalaxyTrakr (https://galaxytrakr.org/) using fasterq_dump v.2.11.0. Paired forward and reverse reads for each SRA collection were assembled using SKESA v.2.3.0 on GalaxyTrakr. Assembled contigs for each isolate were downloaded and then run through translated nucleotide search using a local installation of AMRFinderPlus v.3.11.4 against Pathogen Detection reference gene catalog v.2023-02-23.1, to produce MicroBIGG-E styled output (Suppl. Table 2) from which possible drug-resistance phenotypes were inferred for each isolate (Table 1).
Predicted antimicrobial resistance phenotypes of bacterial isolates from various aquatic animals.
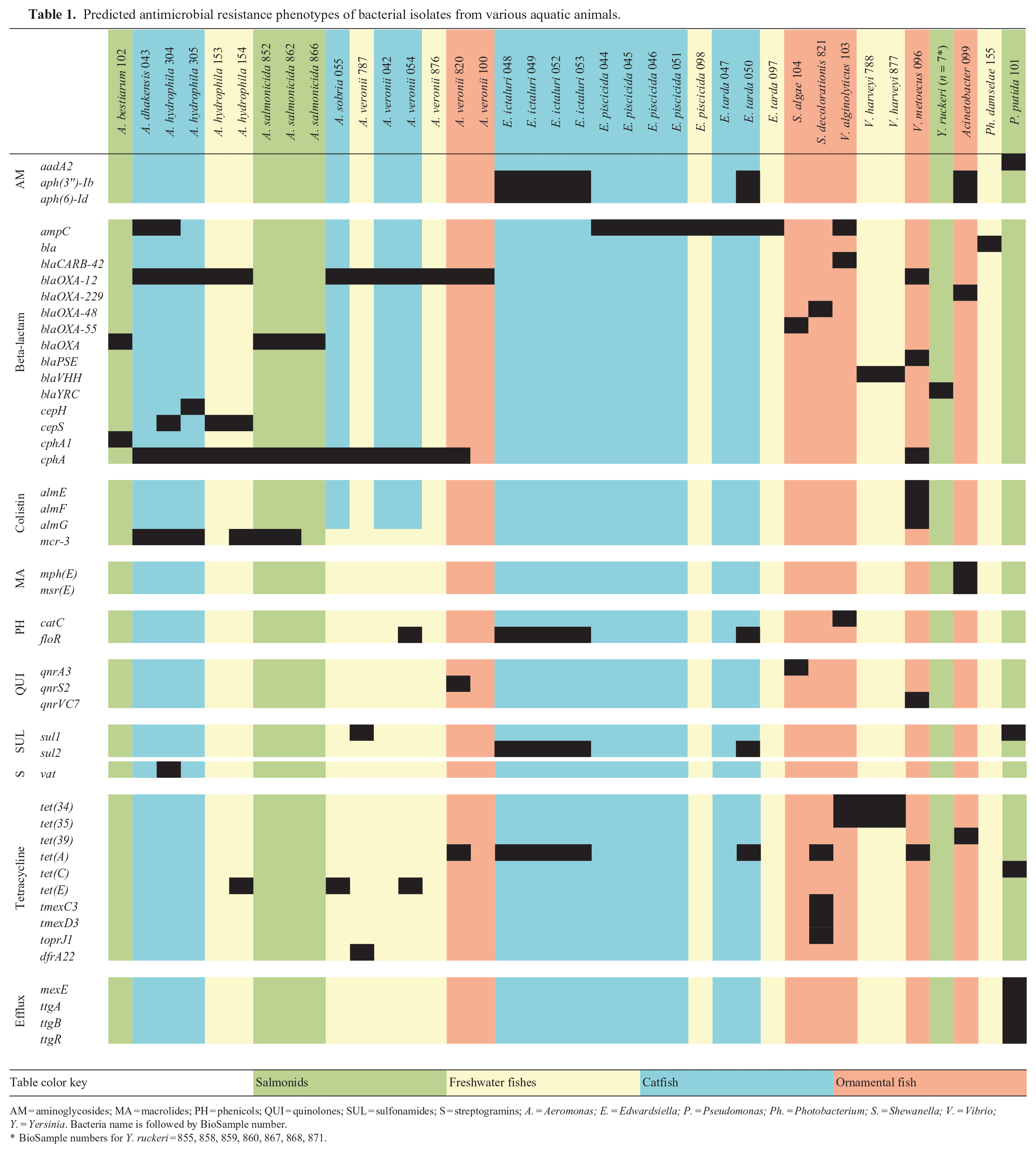
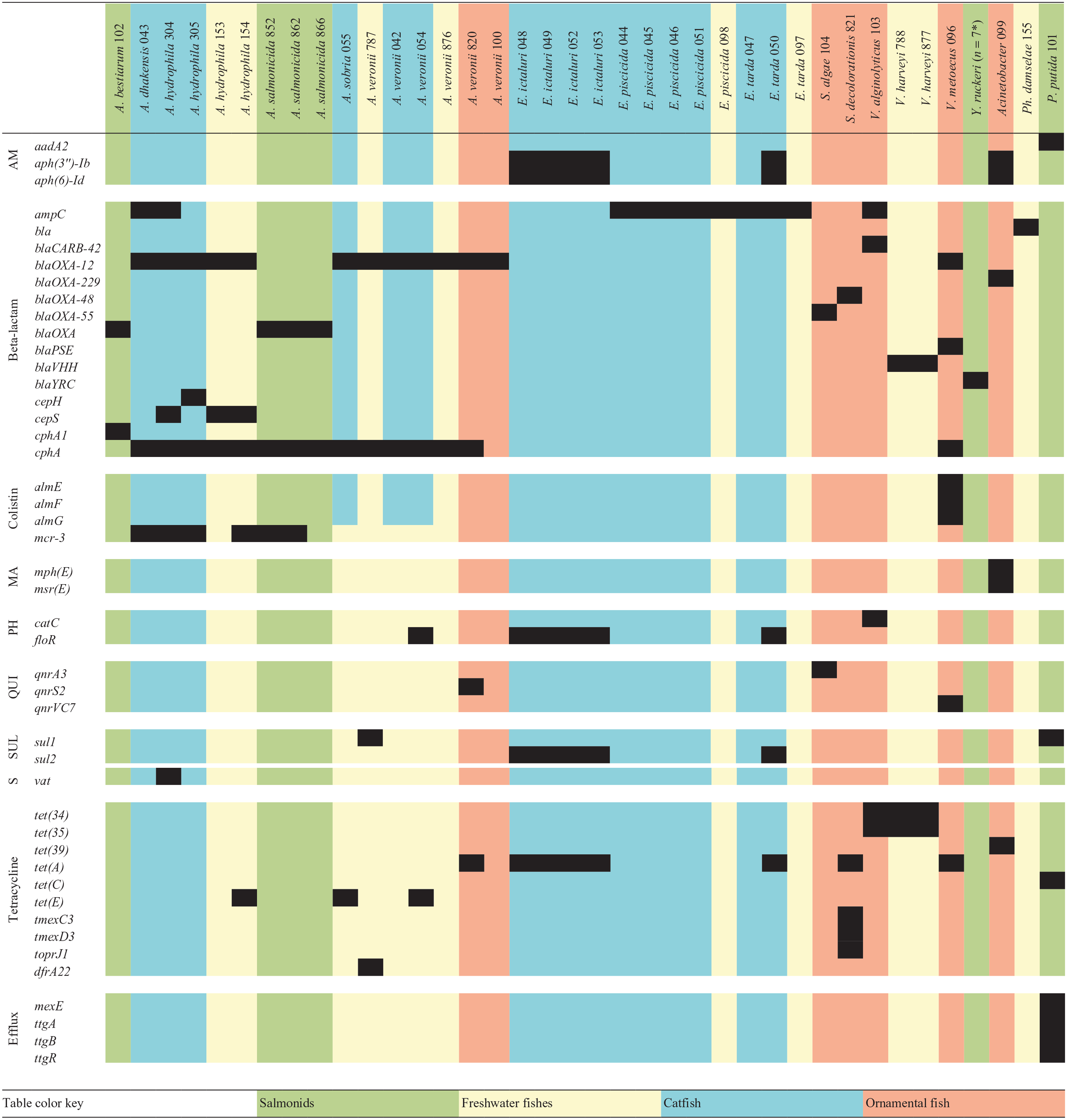
AM = aminoglycosides; MA = macrolides; PH = phenicols; QUI = quinolones; SUL = sulfonamides; S = streptogramins; A. = Aeromonas; E. = Edwardsiella; P. = Pseudomonas; Ph. = Photobacterium; S. = Shewanella; V. = Vibrio; Y. = Yersinia. Bacteria name is followed by BioSample number.
BioSample numbers for Y. ruckeri = 855, 858, 859, 860, 867, 868, 871.
The sequenced isolates comprised 10 genera, including the gram-negative bacteria Aeromonas, Flavobacterium, Edwardsiella, Yersinia, Vibrio, Shewanella, Photobacterium, Pseudomonas, and Acinetobacter, and gram-positive bacteria, Streptococcus. Differences were noted in the bacteria isolated from the various fish species. Aeromonas or Edwardsiella were isolated from catfish, and Aeromonas, Flavobacterium, Pseudomonas, and Yersinia from salmonids. Isolates obtained from other freshwater fishes included Aeromonas, Edwardsiella, Photobacterium, Streptococcus, and Vibrio. Aeromonas, Shewanella, and Vibrio were found in ornamental fish.
Of the 61 isolates analyzed, 17 (28%) were predicted to be pan-susceptible, lacking any canonical ARGs, including Flavobacterium psychrophilum (n = 14), Streptococcus iniae (n = 2), and S. agalactiae (n = 1). As expected, multiple ARGs were identified across the species examined, consistent with the hypothesis that bacteria of aquatic ecosystem origin contain a wealth of AMR determinants.7,14 We identified 19 unique drug-resistance profiles that contain combinations of presumed intrinsic and acquired drug-resistance alleles (Table 1). Inferred AMR phenotypes were based on AMRFinderPlus “Class” or “Subclass” designations given to found alleles, where “Class” describes the broad group or class of antimicrobials to which the element is thought to contribute or confer resistance, and “Subclass” is a non-exhaustive list of specific antimicrobials, resistance to which the element has previously been associated. 10
Of note, beta-lactam resistance genes were observed in many of the gram-negative bacteria, totaling 40 (65.6%) of the isolates sequenced, some with multiple genes present. Genes included blaOXA-12, -48, -55 , and -229 , all encoding for class D beta-lactamase enzymes, of which blaoxa-48, which encodes a carbapenemase, is the most well-known and clinically relevant gene. Interestingly, a 2018 analysis identified Shewanella spp. as the origin for the blaoxa-48 gene which was horizontally transferred to members of the Enterobacterales, including Escherichia coli and Klebsiella pneumoniae. 23 ampC, a chromosomally encoded class C beta-lactamase gene, was present in Aeromonas dhakensis, all Edwardsiella piscicida and E. tarda, and Vibrio parahaemolyticus, consistent with previous reports.6,13 Additionally, ampC-like genes were observed in Aeromonas spp. (cepA, cepH) and Yersinia ruckeri (blayrc ). ampC genes are considered to encode for cephalosporinase enzymes with activity against penicillins, beta-lactam combination agents, and 1st- and 2nd-generation cephalosporins. However, these genes are not always expressed and often require promoter mutations to result in phenotypic beta-lactam resistance. 13 cphA/A1 genes, which encode class B metallo-beta-lactamases, were present in most Aeromonas spp. Last, blaVHH and blaCARB-42 genes, both class A, were found in Vibrio harveyi and V. alginolyticus, respectively. All beta-lactam genes discussed are consistent with intrinsic resistance, being present in all members of the species or genus regardless of previous antimicrobial exposure and unrelated to chromosomal mutation or the horizontal acquisition of new genetic material.7,18 The clinical significance of these genes has yet to be determined.
Genes associated with resistance to florfenicol, ormetaprim-sulfa, and oxytetracycline (floR, catC, sul1/2, and tetA/C/E, respectively), which comprise the only approved drugs for use in aquatic animals in the United States, are predicted to represent acquired resistance. floR was found in Aeromonas veronii (1 of 6), E. tarda (1 of 3), and E. ictaluri (4 of 4); catC was found in V. parahaemolyticus (1 of 1). sul1 was found in A. veronii (1 of 6) and Pseudomonas putida (1 of 1). sul2 was found in Acinetobacter sp. (1 of 1), E. tarda (1 of 3), and E. ictaluri (4 of 4). tet genes were found in Aeromonas hydrophila (1 of 4), A. sobria (1 of 1), A. veronii (1 of 6), E. tarda (1 of 3), and E. ictaluri (4 of 4). Significantly, 1 E. tarda and all E. ictaluri isolates harbored a plasmid contributing resistance to all 3 drugs of importance, a true multi-drug resistance, which is highly concerning. 1
Other notable resistance genes including mcr-3 and mcr-3–like genes, which encode colistin resistance, were found in A. hydrophila (4 of 4), A. salmonicida (2 of 3), and A. dhakensis (1 of 1). mcr-3, and variants of mcr-3, are commonly reported in multiple Aeromonas spp., and despite the high prevalence in the genus, this finding has not been considered intrinsic resistance but is thought to be a reservoir for spread to other clinically relevant bacteria.20,24 Phenotypic resistance is reported but is variably detected in Aeromonas spp. 25
Eighteen isolates harbored genes predicted to confer tolerance to biocides and metals, or loci encoding resistance-nodulation-division (RND)-type efflux pumps. These stress genotypes were limited to 6 species (Table 2). Analysis of Shewanella decolorationis revealed 7 alleles that confer mercury and organomercury resistance, along with RND transporters (Suppl. Table 2). Each Y. ruckeri sequenced harbored the yfeB and ymoA virulence factor genes, which encode the iron/manganese ABC transporter ATP-binding protein YfeB and expression modulating protein YmoA, respectively. 2 Co-expression of these 2 genes is hypothesized to regulate bacterial stress responses and modulate metabolic equilibrium as described for other Yersinia spp.2,17
Stress tolerance haplotypes and associated drug-resistance profiles by organism.
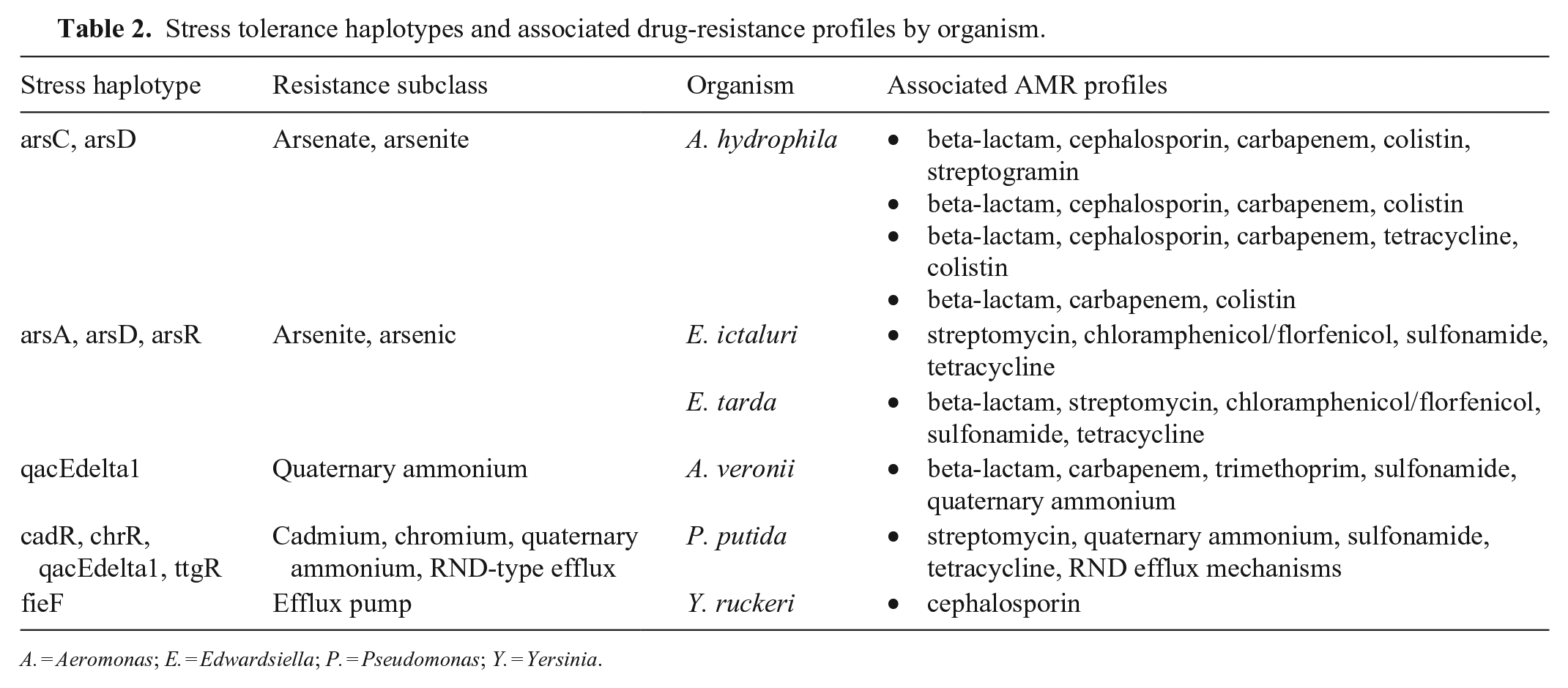
A. = Aeromonas; E. = Edwardsiella; P. = Pseudomonas; Y. = Yersinia.
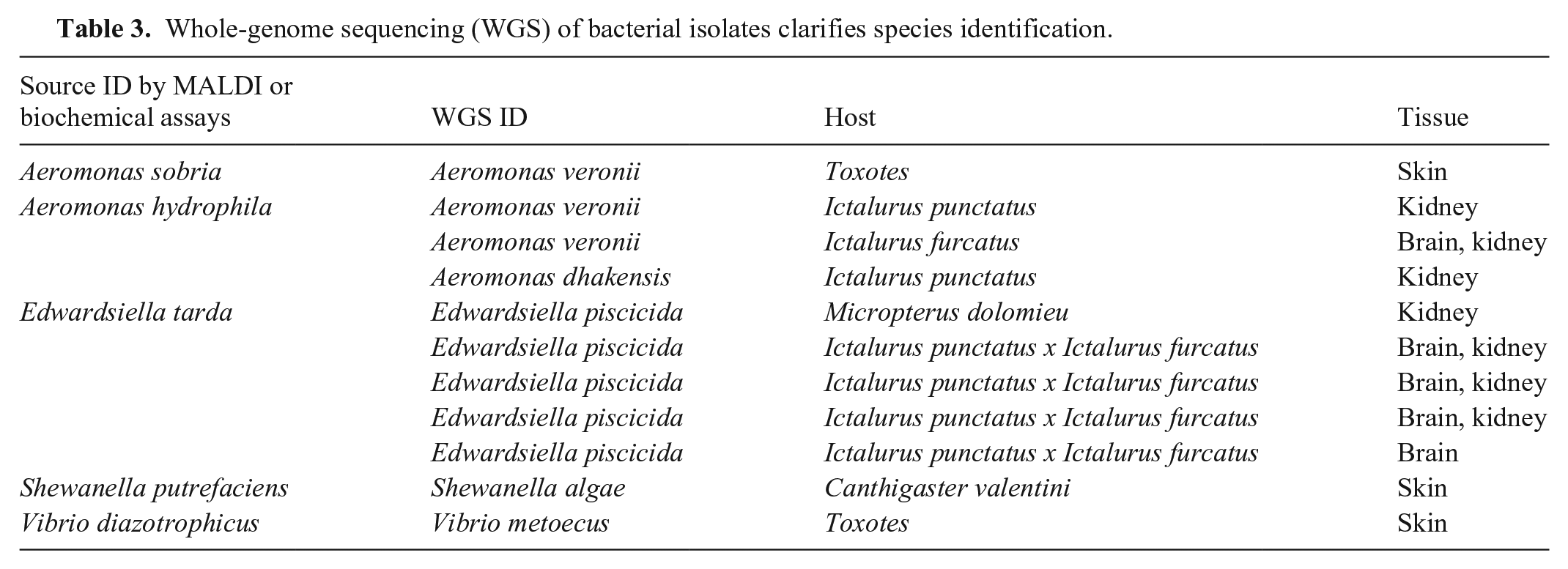
Of 61 isolates, 11 were incorrectly identified relying on MALDI-TOF MS and biochemical assays (Table 3). Assembled genomes were analyzed using PubMLST in GalaxyTrakr, in which ≥ 7 genes are used to identify an isolate. E. piscicida (n = 5), reported as E. tarda, was the most misidentified, followed by multiple Aeromonas spp. (n = 4), which were commonly reported as A. hydrophila.3,9,12 WGS also found discordance between genotypic and phenotypic identities of Shewanella algae and Vibrio metoecus, which were suspected to be S. putrefaciens and V. diazotrophicus, respectively, using traditional methods.
Whole-genome sequencing (WGS) of bacterial isolates clarifies species identification.
Our results contribute to the knowledge gap regarding AMR genotypes of bacteria recovered from various fish species. The identification of ARGs supports the continued need for AMR characterization and monitoring of aquatic animal bacterial pathogens for the purposes of understanding the diversity as well as the scope of resistance to improve treatment outcomes and fish health, safeguard the aquaculture food supply, and protect the public’s health from potential ARG transfer. Interestingly, the discordance between WGS and MALDI-TOF MS bacterial identification highlights the limitations of MALDI-TOF MS together with biochemical methods for correct identification of aquatic bacterial species, and the need for improved methods, including expanded MALDI-TOF MS database libraries to provide greater coverage of these bacteria. Further studies to understand the complexities and factors influencing AMR transfer from aquatic bacteria into clinically significant terrestrial bacteria and acquisition of new ARGs by aquatic pathogens are critical for the prevention of AMR, as well as aquatic animal antimicrobial stewardship.
Supplemental Material
sj-pdf-1-vdi-10.1177_10406387241241042 – Supplemental material for Genomic characterization of antimicrobial resistance in 61 aquatic bacterial isolates
Supplemental material, sj-pdf-1-vdi-10.1177_10406387241241042 for Genomic characterization of antimicrobial resistance in 61 aquatic bacterial isolates by Chrissy D. Eckstrand, Brandi K. Torrevillas, Rebecca M. Wolking, Marla Francis, Laura B. Goodman, Olgica Ceric, Trevor L. Alexander, Kevin R. Snekvik and Claire R. Burbick in Journal of Veterinary Diagnostic Investigation
Footnotes
Acknowledgements
We acknowledge the assistance of the personnel at the Florida Department of Agriculture and Consumer Services–Bronson Animal Disease Diagnostic Laboratory, North Carolina State University–Veterinary Microbiology and Molecular Diagnostic Laboratory, Mississippi State University–Aquatic Research and Diagnostic Laboratory, the Cornell University–Animal Health Diagnostic Center, and the Washington State University–Washington Animal Disease Diagnostic Laboratory.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to research, authorship, and/or publication of this article. The views expressed in this manuscript are those of the authors and may not reflect the official policy of the Department of Health and Human Services, the U.S. Food and Drug Administration, or the U.S. Government.
Funding
This project was funded by grant 5U18FD006453 from the Food and Drug Administration, Center for Veterinary Medicine, Veterinary Laboratory Investigation and Response Network (Vet-LIRN).
ORCID iDs
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.