
Abstract
Herpesviruses are associated with disease in many penguin species. Herpesvirus-associated lesions can cause significant morbidity and mortality in penguins and have been identified in African penguins (Spheniscus demersus), Humboldt penguins (Spheniscus humboldti), and a little blue penguin (Eudyptula minor) infected with spheniscid alphaherpesvirus 1 (SpAHV1). Further investigation is necessary to understand the impact of herpesviruses on penguin health, but there are no rapid, sensitive, and specific methods for detecting and quantifying herpesviral load. We therefore developed a quantitative real-time PCR (qPCR) assay for the detection of SpAHV1 in penguins. TaqMan primer-probes targeting the DNA polymerase gene were designed using a commercial software program. Inter- and intra-assay variability, dynamic range, limit of detection, and analytical specificity were assessed. We used our assay to analyze previously collected field samples from Punta San Juan, Peru, in which conventional consensus PCR had detected one SpAHV1-positive penguin sample. Our qPCR assay was highly specific for SpAHV1. It had a dynamic range of 107–101 target copies per reaction and performed with high efficiency and low intra- and inter-assay variability. Reaction efficiency was not impacted by penguin DNA from SpAHV1-negative tracheal swabs. We detected an additional field sample as positive with our newly developed qPCR assay, and although this likely represents detection of another infected penguin, the true disease status of this population is currently uncharacterized given that no gold-standard test exists for SpAHV1. Our qPCR assay may provide a valuable tool in the surveillance and characterization of SpAHV1 in penguins.
Herpesviruses are double-stranded DNA viruses that infect a wide range of vertebrate hosts, including birds, mammals, amphibians, and reptiles along with a few invertebrate species. 5 The clinical consequences of herpesviral infections can range from latent infections with no clinical signs to severe and sometimes fatal disease. Occurrence and severity of herpesviral infection depends on the host immune status, host–virus co-evolutionary history, and comorbidities.9,14,16
Numerous herpesviruses have been detected in taxonomically diverse avian families; however, only 8 avian herpesvirus species in the genera Iltovirus and Mardivirus are currently recognized by the International Committee on Taxonomy of Viruses (ICTV; https://ictv.global/taxonomy): gallid alphaherpesvirus 1 (GaAHV1; syn. infectious laryngotracheitis virus; Iltovirus gallidalpha1), gallid alphaherpesvirus 2 (GaAHV2; syn. Marek disease virus; Mardivirus gallidalpha2), gallid alphaherpesvirus 3 (GaAHV3; syn. gallid herpesvirus 3; Mardivirus gallidalpha3), meleagrid alphaherpesvirus 1 (MeAHV1; syn. turkey herpesvirus; Mardivirus meleagridalpha1), anatid alphaherpesvirus 1 (AnAHV1; syn. duck enteritis virus; Mardivirus anatidalpha1), psittacid alphaherpesvirus 1 (syn. Pacheco disease virus; Iltovirus psittacidalpha1), columbid alphaherpesvirus 1 (CoAHV1; syn. pigeon herpesvirus; Mardivirus col-umbidalpha1), and spheniscid alphaherpesvirus 1 (SpAHV1; Mardivirus spheniscidalpha1) from penguins. 7 Many other avian herpesviruses have not been officially classified given the lack of genetic sequencing data, and this includes most herpesviruses infecting non-domestic birds.6,9,10,12 Several of these herpesviruses have been associated with outbreaks and mortality events in managed-care and free-living settings (e.g., gruid herpesvirus 1, 9 Fregata magnificens herpesvirus 1 [FmagHV1] 6 ) underscoring the potential importance of herpesviruses for avi-an conservation.
At least 3 genetically distinct herpesviruses have been identified in penguins (Sphenisciformes, Spheniscidae): magellanic penguin herpesviruses 1 and 2 (MPHV1, MPHV2) 12 and SpAHV1. 16 MPHV1 was associated with severe, hemorrhagic respiratory disease in 98 oiled juvenile Magellanic penguins (Spheniscus magellanicus) presented to a wildlife rehabilitation center in Brazil in June–July 2011. MPHV2 was detected in 5 of 31 (16%) nestling and 4 of 142 (2.8%) adult Magellanic penguins sampled at 4 nesting sites in Argentina during January 2014. All sampled birds were apparently healthy with no clinical signs of illness. 12
SpAHV1 was originally isolated in culture, and the complete genome was sequenced from infected juvenile Humboldt (Spheniscus humboldti) and African (Spheniscus demersus) penguins kept in German zoos suffering from diphtheroid oropharyngitis or laryngotracheitis and necrotizing enteritis.13,14 This constellation of lesions is known as penguin diphtheria, and is one of the most important causes of morbidity and mortality in wild and managed-care juvenile penguins. 1 Although Koch postulates have not been fulfilled for SpAHV1 as a causative agent for penguin diphtheria, further investigation into the role of SpAHV1 in penguin health is clearly indicated. For example, SpAHV1 has also been detected in a hatchling little blue penguin (Eudyptula minor) with encephalitis, demonstrating that this virus may be clinically important for a variety of penguin species. 16
Virus isolation and conventional consensus PCR have been used to detect SpAHV1; however, the conventional consensus PCR assay used to detect SpAHV1 has a variable limit of detection (LOD) depending on the herpesvirus. 14 Additionally, this assay is not specific for SpAHV1, and DNA sequencing is required to confirm the identity of positive PCR products. Quantitative real-time PCR (qPCR) has not been reported for the detection of SpAHV1 in penguins, but could provide improved speed, sensitivity, specificity, and efficiency compared to conventional consensus PCR. A validated qPCR assay for SpAHV1 could allow identification of carrier birds and therapeutic monitoring of clinically affected penguins. Given the latent character of herpesviruses, early detection of recurrence and viral shedding is critical for informing disease management decisions.
We developed a TaqMan qPCR assay for detecting SpAHV1 in penguins. We hypothesized that a qPCR hydrolysis probe–based assay would have high analytical sensitivity and specificity for detecting SpAHV1 in penguins. Our assay should provide a beneficial tool for the detection and epidemiologic characterization of SpAHV1.
Materials and methods
Plasmid preparation and standard curve generation
We selected the SpAHV1 DNA polymerase gene as a target for qPCR primer-probe design because it is highly conserved among available SpAHV1 whole-genome sequences and because DNA polymerase sequences for several other avian herpesviruses were also available in GenBank to facilitate specificity analyses. A 1,080-bp segment of the SpAHV1 DNA polymerase gene was retrieved from GenBank, 14 synthesized (GeneArt; Thermo Fisher), and inserted into a pMA-RQ (ampicillin resistance) plasmid (Integrated DNA Technologies). This plasmid was transformed into Escherichia coli using the TOPO TA cloning kit (Invitrogen). Bacterial selection was attained using lysogeny broth agar plates (Fisher Bioreagents) containing 100 μg/mL of ampicillin; plasmid DNA was then isolated from overnight bacterial cultures (QIAfilter plasmid Maxi kit; Qiagen). The presence of the SpAHV1 insert was verified by sequencing (ACGT). Plasmids were linearized by restriction enzyme digestion (PvuII; New England Biolabs) and visually confirmed on 1% agarose gel. Phenol–chloroform extraction and ethanol precipitation were used to remove the restriction enzyme reaction components, and DNA purity and quantity were established (Nanodrop spectrophotometer; Thermo Scientific).
Target DNA copy number was calculated based on a DNA length of 3,421 bp (1,080-bp insert; 2,341-bp plasmid backbone) and a DNA concentration of 1,322 ng/μL for our specific plasmid stock solution. This linearized stock plasmid was then diluted to a concentration of 4.00 × 108 target copies/µL using sterile water. Following this, nine 10-fold serial dilutions were made from 4.00 × 107 to 4.00 × 10–1 target copies/μL by mixing 50 µL of the preceding dilution with 450 µL of sterile water. Dilutions from 4.00 × 106 to 4.00 × 10–1 target copies/µL, which corresponded to 1.00 × 107 to 4.00 × 100 target copies per reaction (2.5 µL of each plasmid dilution was used per qPCR reaction), were used to generate the standard curve.
Real-time qPCR assay
We followed the minimum information for publication of quantitative real-time PCR experiments (MIQE) guidelines. 4 We tested 2 TaqMan-MGB FAM dye labeled (Applied Biosystems [ABI] TaqMan primers; Thermo Fisher) qPCR assays: SpAH-V1-A and SpAHV1-B. Primers for each assay were designed using a commercial software program11,17 based on the seque-nce of the SpAHV1 DNA polymerase gene segment in our clone. The SpAHV1-A TaqMan assay was performed using a forw-ard primer (5′-ACTGGATCTATGTGAAACCCGG-3′), reverse primer (5′-ACAAAGCGTGTCTGGCAAAG-3′), and probe (5′-CCCTGGTAACCAACAACCCTGCCCCCTGCA-3′), targeting a 105-bp segment within the DNA polymerase catalytic subunit (GenPept locus YP_009342373, bp coordinates 1,563–1,668). The SpaAHV1-B TaqMan assay was performed using a forward primer (5′-CCCTTCATTAGCATTTTTCCCCC-3′), reverse primer (5′-TAGGTGATGCGATGGCGAAG-3′), and probe (5′-TGGCGCTTTGAACAGGTCTGCGGTGATGCG-3′), targeting a 148-bp segment of the DNA polymerase catalytic subunit (GenPept locus YP_009342373, bp coordinates 833–981). Primer BLAST was performed to confirm in silico specificity of primer design. 20
qPCR assays were performed using a real-time PCR thermocycler (ABI QuantStudio 3 real-time PCR system; Thermo Fisher), and data were analyzed (QuantStudio design and analysis software v.1.5.2). Each reaction contained 12.5 μL of 20 × master mix (TaqMan Platinum PCR Supermix-UDG with ROX; Invitrogen), 1.25 μL of 20 × TaqMan primer-probe (final concentrations in each PCR reaction: 900 nM forward primer, 900 nM reverse primer, and 250 nM probe), 2.5 μL of plasmid dilution, DNA sample, or non-template control (sterile water), and 8.75 μL of sterile water to a final volume of 25 μL. Reactions were loaded into 96-well reaction plates (ABI MicroAmp optical; ThermoFisher) and sealed with clear adhesive film (ABI MicroAmp). The qPCR cycling parameters were as follows: 1 cycle at 50°C for 2 min, followed by 40 cycles of 95°C for 15 s and 60°C for 60 s, then a final cycle of 72°C for 10 min.
Analytical specificity
Analytical specificity of the SpAHV1-A and SpAHV1-B assays was evaluated using synthetically produced DNA (GeneArt) based on the DNA polymerase gene sequences from 15 closely related avian herpesviruses: CoAHV1 (GenBank GQ478232), AnAHV1 (AF064639), FmagHV1 (EU867220), gaviid HV1 (GU130289), thalassarchid HV1 (KR092313), sulid HV1 (KP003804), vulture HV1 (AY571867), neotropic cormorant HV1 (KY769944), ciconiid HV1 (MN017363), penguin HV1 (KT006357), and penguin HV2 (KR338839). DNA from pure cultures of GaAHV1, GaAHV2, GaAHV3, and MeAHV1 was also used to assess the specificity of the SpAHV1 assays.
Limit of detection, repeatability, dynamic range, and efficiency in sample matrix
LOD was determined by testing 5 technical replicates of a plasmid dilution series ranging from 4.00 × 106 to 4.00 × 10–1 target copies/µL (corresponding to 1.00 × 107 to 1.00 × 100 target copies per reaction). LOD was conservatively set as the lowest standard dilution in which 95% of replicates successfully amplified. Inter-assay variability was evaluated between 2 duplicate plates containing 5 replicates of the above-described standard curve; intra-assay variability was determined by comparing 2 identical dilution series (5 replicates each) on the same plate. Inter- and intra-assay variation were determined from the mean Cq, SD, and CV of each SpAHV1 plasmid DNA dilution (4.00 × 106 to 4.00 × 10–1 target copies/µL, corresponding to 107–100 copies per reaction).
Efficiency curves were performed using DNA extracted from SpAHV1-negative penguin tracheal swabs spiked with plasmid dilutions (4.00 × 106 to 4.00 × 10–1 target copies/µL, corresponding to 107–100 copies per reaction). These were used to determine whether assay performance was impacted by tracheal swab sample matrix. Tracheal swabs were sourced from a managed-care population of apparently healthy Humboldt penguins that were being sampled for routine pathogen surveillance. SpAHV1-negative status for these tracheal swabs was determined using consensus herpesviral PCR, with DNA extraction and PCR methods following publications. 2
Method comparison
We compared the performance of our SpAHV1-A assay to the conventional consensus herpesviral PCR assay by testing 62 tracheal swab DNA samples from a free-ranging Humboldt penguin population within the 60-ha Punta San Juan Marine Protected Area (15°22′S, 75°11′O), Ica, Peru. Details of the collection, DNA extraction, and conventional consensus herpesvirus PCR results for these samples have been reported. 2 Quantitative PCR was performed using the SpAHV1-A primer-probe and the reaction conditions described above. Each sample, standard (107–100 copies per reaction), and non-template control were assessed in triplicate. Resulting SpAHV1 quantities (target copies/μL) were normalized based on starting DNA concentration (as determined by spectrophotometry) and reported as target copies/ng DNA.
Results
Analytical specificity
The SpAHV1-B assay amplified DNA from CoAHV1, which is closely related to SpAHV1; the SpAHV1-A assay did not. Neither assay amplified the other 14 avian herpesviruses tested. Given its increased analytical specificity, we evaluated intra-assay variability, inter-assay variability, and efficiency only for the SpAHV1-A assay.
Assay characteristics, dynamic range, limit of detection, repeatability, and efficiency in sample matrix
Serial 10-fold dilutions of positive control plasmids were performed for TaqMan qPCR assay SpAHV1-A, and amplification plots and standard curves based on Cq values were generated based on these dilutions. The dynamic range for SpAHV1-A was 1.00 × 107 to 1.00 × 101 target copies per reaction with an R2 of 0.997 and slope of −3.37 (Fig. 1). LOD was 101 target copies per reaction, although 100 target copies per reaction were detected in the intra-assay plate run. Intra-assay CVs for SpAHV1-A were 0.15–4.17%; inter-assay CVs were 0.18–1.97%. The inter-assay variability run was unable to reliably detect SpAHV1-A at 100 target copies per reaction (Table 1).
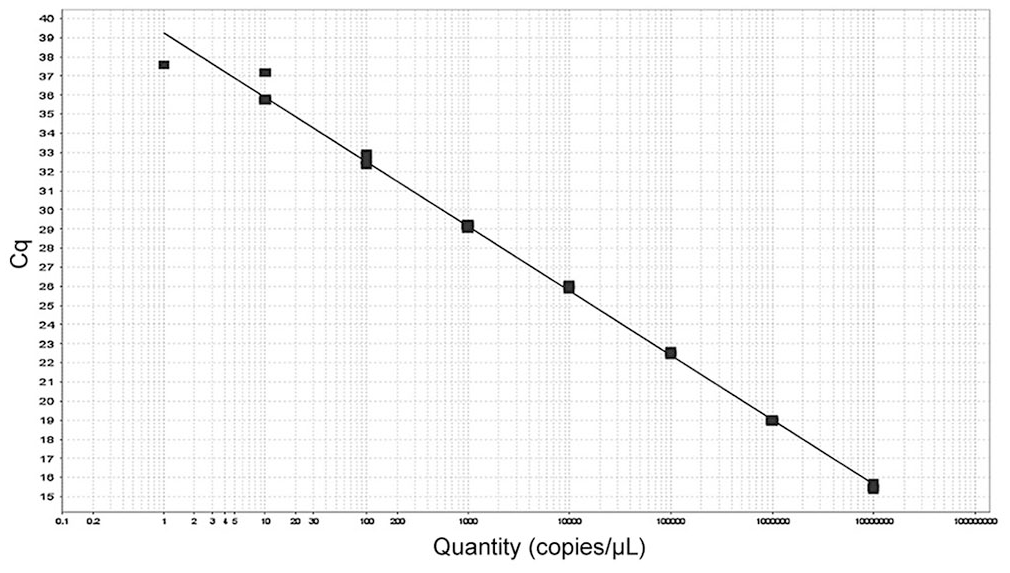
Standard curve for SpAHV1 assay. Target = target 1; slope = –3.37; y-intercept = 39.3; R2 = 0.997; efficiency = 98.1%.
Intra- and inter-assay variability in Cq values for a TaqMan qPCR SpAHV1-A assay using an 8-point standard curve from 107–100 SpAHV1 target copies per reaction.
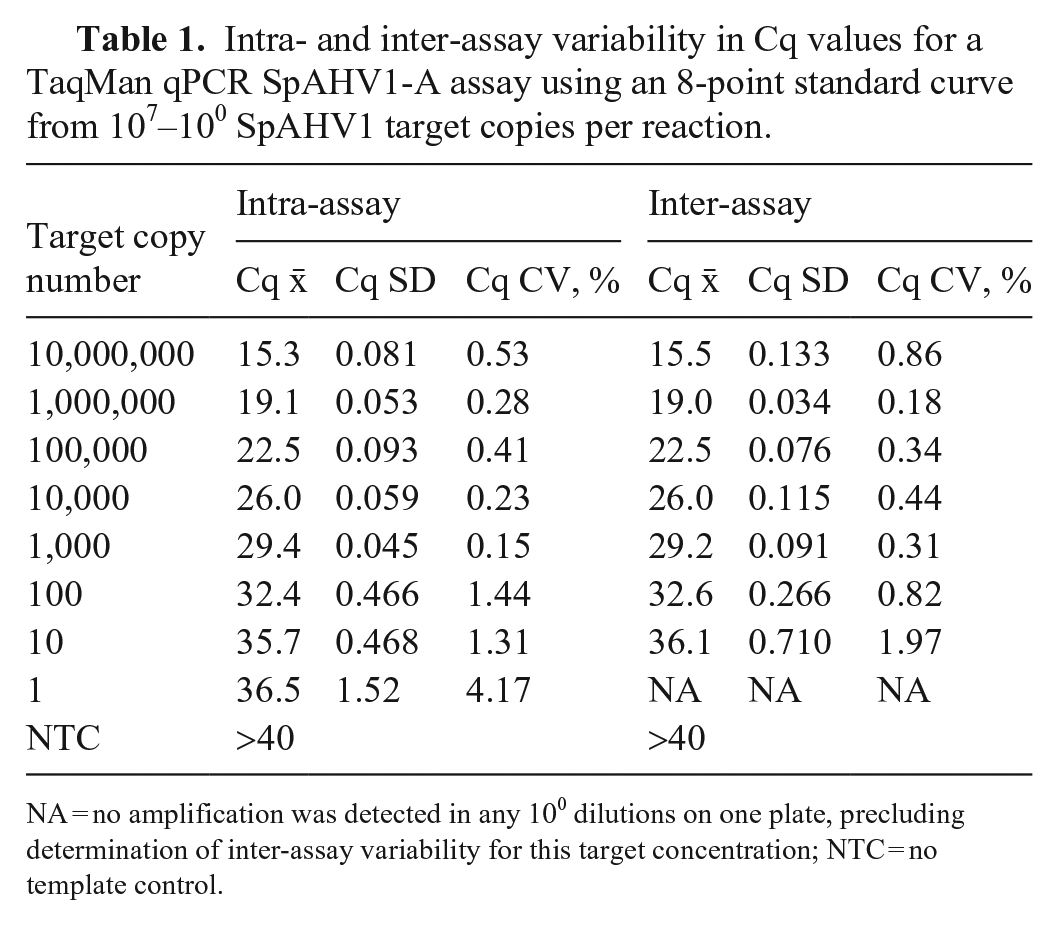
NA = no amplification was detected in any 100 dilutions on one plate, precluding determination of inter-assay variability for this target concentration; NTC = no template control.
Efficiency curves performed with SpAHV1-negative tracheal swab DNA spiked with plasmid dilutions revealed no significant efficiency loss in the presence of penguin tracheal swab DNA. Compared to the standard curve derived from plasmid dilutions (slope −3.37, R2 = 0.997, efficiency 98.1%), plasmid dilutions spiked into tracheal swab DNA (slope −3.43, R2 = 0.999, efficiency 95.7%) had comparable and acceptable performance. A standard curve slope between −3.1 and −3.6 (90–110% efficiency) with a correlation coefficient (R2) > 0.98 is considered acceptable for most detection qPCR assays. 3 Our SpAHV1-A assay achieved levels of efficiency and variation within these acceptable ranges.
Method comparison
The SpAHV1-A qPCR assay detected SpAHV1 DNA in 2 of 62 (3.2%; 95% CI: 0.9–11.0%) tracheal swab DNA samples from Humboldt penguins in Peru. The previously reported SpAHV1-positive adult was also confirmed positive through our SpAHV1-A qPCR assay, with 82.8 target copies/ng DNA. This animal was a 4.5-kg male with no clinically relevant physical examination abnormalities. The new positive animal was an adult, 3.9-kg female with no recorded clinical signs that was sampled in 2016 and had 0.25 SpAHV1 target copies/ng DNA. Additional testing to confirm true disease status was not available for any sample.
Discussion
Developing assays for wildlife disease surveillance is challenging in many respects. One of the more significant hurdles involved with evaluating the analytical specificity of the SpAHV1 qPCR primer-probes was the use of synthetically produced DNA for several avian herpesviruses. Inclusion of pure culture material or biologic samples collected from herpesvirus-infected birds would have been preferable because this would have allowed for a higher degree of confidence in determining the analytical specificity of our primer-probes. However, logistic challenges, including lack of sample availability and/or permitting necessary for international sample movement, precluded the incorporation of these materials into our study. Future work can and should be performed using biomaterials from these and other herpesviruses to confirm the analytical specificity of our SpAHV1-A qPCR assay.
Although our assay has been partially analytically validated, there are additional limitations in validating its detection use. Our study lacks the minimum number of positive reference samples to determine diagnostic sensitivity according to established guidelines. 18 At least 30 positive samples are recommended to evaluate the diagnostic performance of new tests in wildlife, but given the low number of reported SpAHV1 cases, this is not an achievable objective. 19 It is also challenging to complete clinical validation for this assay because there is no gold standard for SpAHV1 testing, and true-positive and true-negative cases are difficult to identify. Therefore, our assay only fulfills the MIQE requirements for analytical sensitivity and specificity, and can only be considered partially validated. The limited availability of penguin clinical samples also restricts the assessment of inter-laboratory reproducibility, an additional component of clinical validation.4,18 These are commonly recognized limitations for assays developed for use in wildlife.8,15,18,19 Despite this, our partially validated SpAHV1-A qPCR assay shows promise; future studies can pursue clinical validation as known-positive and known-negative samples become available.
Although we could not complete true clinical validation of our SpAHV1-A qPCR assay at this time, comparison to an existing SpAHV1 detection method was pursued to provide some limited initial data on assay performance. Results of the qPCR and consensus PCR methods were in agreement for 61 of 62 tracheal swab DNA samples; however, one sample tested positive via qPCR and negative via consensus PCR. Although this likely represents detection of an additional SpAHV1-infected penguin (or detection of SpAHV1 nucleic acid present on the swab), the copy number of this sample was low and sequence confirmation could not be obtained. It is also possible that this positive test result represents nonspecific amplification. Samples for confirmatory testing, such as virus isolation, are not available from this population, and the true disease status of the birds is unknown. Further studies are indicated to identify a gold-standard method for detecting SpAHV1, determine the diagnostic sensitivity and specificity of available testing methods, and complete analytical and clinical validation of existing assays.
Future studies on the performance of the SpAHV1-A qPCR should also examine the effects of sample quality and handling, given that these pre-analytical factors can negatively impact test accuracy, especially from field-collected samples. Test performance may also vary over the course of infection, and host–pathogen biologic factors, such as shedding patterns and coinfections, can also impact test accuracy. Further research will be necessary to quantify the effects that these factors may have on the performance of our SpAHV1-A qPCR.
Footnotes
Acknowledgements
We thank Keith Jarosinski of the University of Illinois Urbana-Champaign Department of Pathobiology for providing gallid alphaherpesvirus 1–3 and meleagrid alphaherpesvirus 1 DNA for specificity testing.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.