
Abstract
Clinical manifestations of leptospirosis are diverse and very similar to other febrile diseases, hence early and accurate detection of subclinical infections is a key element in disease control. We evaluated immunomagnetic separation (IMS) capture technology coupled with a standard quantitative PCR (qPCR) system for the detection of pathogenic Leptospira in urine samples from 803 cows from dairy herds with a history of clinical cases of leptospirosis. The urine samples were first processed in a purification step, then subdivided into 2 subsamples, one that continued to DNA extraction and direct qPCR, and one that was pretreated by IMS before continuing to DNA extraction and qPCR. Overall, 133 of 803 (16.6%) samples were IMS-qPCR positive, whereas only 92 of 803 (11.5%) were positive when using direct qPCR. Statistically significant differences were observed between the mean estimated Leptospira load between the IMS-qPCR and the direct qPCR positive urine samples. The IMS-qPCR technology revealed a larger number of positive results and higher bacterial loads than direct qPCR. This difference is most likely the result of the high antigen-binding capacity and capture efficiency of the IMS system. The use of polyclonal antibodies produced by the inoculation of 3 synthetic peptides, which make up the extracellular regions of the LipL32 protein, provided a high detection capacity to the IMS-qPCR technique, resulting in performance superior to direct qPCR.
Introduction
Leptospirosis, a bacterial zoonotic disease with a worldwide distribution, is caused by spirochetes of the genus Leptospira and encompasses a wide spectrum of clinical diseases in humans, including multi-organ failure with a high mortality rate. 10 Farming activities were recognized as an important risk factor before animal host species were identified. 1 Rodents were first identified as a potential source of human infection, followed by dogs.2,4 The role of livestock as reservoirs was not determined until several decades later. 4 Frequently, abortion or stillbirth is the only clinical sign detected in adult cattle infected by pathogenic Leptospira. 8 Infected calves usually develop a severe, acute form of the disease (with clinical signs such as fever, jaundice, and hematuria) that is frequently fatal. 2
Because of the diversity of clinical signs, the detection of pathogenic Leptospira is difficult and depends upon a variety of laboratory assays such as the detection of specific antibodies by microscopic agglutination test, indirect hemagglutination assay, or ELISA. In addition, Leptospira or their components may be detected in urine or tissues by culture, dark field microscopy, immunostaining, or PCR.1,4,6
Immunomagnetic separation (IMS) has been reported to be used for Leptospira detection. 5 This concentration method can be coupled with any test for Leptospira detection regardless of whether it is based on antigens, genes, phage binding, or growth in culture media. If effective, IMS can provide cleaner samples (i.e., free of contaminating microbes or PCR inhibitors) and a higher yield of Leptospira (i.e., improved analytical sensitivity) via a one-step, low-cost procedure. 5
The combination of IMS and PCR increases both test specificity and sensitivity.13,17,18 There remain some hurdles to widespread use of the IMS-PCR technology for Leptospira detection in veterinary specimens. The analytical sensitivity of the IMS method has been evaluated with only a small number of Leptospira strains and under experimental conditions. 5 We describe herein the development, optimization, and analytical evaluation of an IMS–quantitative PCR (IMS-qPCR) protocol to facilitate the detection of pathogenic Leptospira from cattle urine samples obtained under field conditions.
Materials and methods
Study population and sample collection
Sampling was conducted among 15 smallholder and 23 medium-size dairy farms located in the Los Ríos and Los Lagos regions of southern Chile, between October 2016 and January 2017. Verbal consent was obtained from all farmers who participated in the study. The smallholder farmers are subsistence farmers who produce < 100,000 L of milk/y, and their cattle graze outside year-round and are fed little or no concentrate. The medium-size herds represent the typical dairy farms of the area in terms of breed (Holstein), herd size (200–500 animals), and management practices (graze in rotational paddocks year-round, milked twice a day, 305-d milk production, 220,000–4,500,000 L/y).
A targeted sampling strategy was used to maximize the likelihood of testing Leptospira-infected cattle by selecting herds with a history of clinical leptospirosis, mainly associated with Pomona and Hardjo serovars. We collected 803 urine samples from adult cattle from the 38 study herds. In order to detect pathogenic Leptospira in each of the samples, urine samples (25 mL) were collected by direct stimulation of the vulvar area. Urine samples were kept at room temperature until they were transferred and processed, on average within 4 h, at the Laboratorio de Enfermedades Infecciosas, Instituto de Medicina Preventiva Veterinaria, Facultad de Ciencias Veterinarias, Universidad Austral de Chile.
Detection of pathogenic Leptospira in urine specimens was conducted through a comparative approach based on both direct qPCR and IMS-qPCR from each urine sample. Each urine sample was centrifuged at 4,000 × g for 15 min; the pellet was then resuspended in 1 mL of phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4 [pH 7]), transferred to a 1.5-mL microcentrifuge tube, and re-centrifuged at 11,000 × g for 5 min. Finally, the supernatant was discarded, and the pellet was resuspended in 1 mL of PBS. After this first cleaning and purification step, the sample was subdivided, with a 100-μL aliquot used for DNA extraction and direct qPCR and another 100-μL aliquot undergoing a pretreatment step of immune separation before proceeding with DNA extraction and direct qPCR.
Polyclonal antibody production
In order to produce polyclonal antibodies against LipL32 (an outer membrane lipoprotein 7 ), 3 peptide chains (EP3, EP4, and EP6), 9 each with a size of ~20 kDa, were chemically synthesized (this mixture hereafter referred to as P2). Sequences of these LipL32 epitopes have been described in silico as the most immunogenic and are found outside the leptospiral bacterial cell wall. 9 A glutaraldehyde cross-linking protocol with hemocyanin carrier protein (Imject Blue carrier protein; Thermo Fisher Scientific) was performed on the peptides, as described previously, 11 with some modifications. Briefly, 10 mg of hemocyanin was incubated with 1 mg of each peptide in the presence of 0.15% glutaraldehyde (MilliporeSigma) at room temperature for 2 h. The reaction was terminated by the addition of 1/10 (v/v) of 1 M glycine, and the sample was dialyzed against 0.1 M borate buffer (18.55 g of boric acid; 2,850 mL of H2O; pH 8.5) for 24 h at 4°C. From this protocol, an emulsion was made with 200 μg of each of the peptides and Freund complete adjuvant (MilliporeSigma), with 1 mL injected subcutaneously in 4 different locations on the back of a 2.5-kg New Zealand male rabbit. The laboratory animal was provided by the Instituto de Salud Pública (Chilean Government). This rabbit was managed in strict accordance with the recommendations in the Guide for Use of Animals for Research of the Universidad Austral de Chile (https://www.uach.cl/organizacion/vicerrectoria-investigacion-desarrollo-y-creacion-artistica/utiles/subcomite-en-uso-de-animales-en-investigacion, Spanish.). The animal was housed in an individual metal cage with free access to food and water. Polyclonal antibodies were obtained from the rabbit serum and stored at −20°C until use. The serum was used directly to perform the IMS without any purification step.
Polyclonal antibody validation
In order to evaluate the binding of the anti-LipL32 antibody to the target protein in pathogenic Leptospira, one strain (107 bacteria/mL) of each of 6 serovars (Table 1) was used. The Leptospira strains were provided by the National Collaborating Centre for Reference and Research on Leptospirosis (Amsterdam, The Netherlands). The cultures were homogenized in the extraction buffer (50 mM Tris, pH 8.0; 1 mM EDTA; 100 mM NaCl; 1 mM phenylmethylsulfonyl fluoride). The Leptospira cells were disrupted by tip sonication at 70% in an ice bath with two 10-s rounds. Samples were quantified (Bradford reagent; MilliporeSigma). Twenty µg of whole protein extract was separated by SDS-PAGE and electro-transferred to a 0.45-µm nitrocellulose membrane (GE Healthcare). Membranes were blocked with 1% bovine serum albumin (BSA) in 0.2% Tween-20 for 1 h at room temperature. Primary antibody (1:1,000 v/w) incubation was performed overnight, using continuous stirring at 4ºC. The following day, membranes were washed in 0.2% Tween-20 solution in Tris-buffered saline (TBS; pH 7.4) and then incubated with anti-rabbit IgG–horseradish peroxidase secondary antibody (1:2,000 v/w; Santa Cruz Biotechnology) for 1 h at room temperature. Finally, the immunoreactive signal was revealed with a solution of luminol–hydrogen peroxide (ECL; Thermo Scientific) and digitally displayed (LI-COR Odyssey Fc system; Biosciences). Primary antibody specificity was evaluated in parallel by the use of primary antibody that had been pre-adsorbed with immunogenic peptides (P2) overnight.
Details of Leptospira strains used in evaluation of an immunomagnetic separation PCR assay to detect pathogenic Leptospira in bovine urine samples.

All reference strains were provided by the National Collaborating Centre for Reference and Research on Leptospirosis (Amsterdam, The Netherlands).
Indirect immunofluorescence
To demonstrate the binding of the anti-LipL32 antibody to the membrane of pathogenic Leptospira, an immunocytochemistry assay was performed using L. interrogans serovar Icterohaemorrhagiae strain Icterohaemorrhagiae (Table 1). Smears of this Leptospira strain were prepared on slides, fixed with 2% paraformaldehyde in 2.5 mM MgCl2, and then blocked with 5% BSA in 0.2% Tween-20 in TBS for 60 min at room temperature. Samples were incubated overnight with the primary antibodies (1:50 v/w) in a moist chamber at 4°C. As a negative control, the primary antibody was not added. In a second control, the primary antibody was pre-absorbed with the immunogenic peptides (P2). Next, incubation with the secondary antibody was performed, with antibodies conjugated to fluorochrome Alexa 488, for 60 min at room temperature. Bacterial DNA was counterstained with 2 µM propidium iodide and washed twice in TBS. Finally, the slides were mounted, and stained cells were visualized and evaluated in an inverted epifluorescence microscope (DMI3000 B; Leica) coupled to a digital camera (DFC 425 C; Leica). The images obtained were processed with Adobe Photoshop 6.0.
Use of coated magnetic beads
Magnetic beads coated with anti-rabbit antibodies (M-280 sheep anti-rabbit IgG; Dynabeads) were used. The 100-µL aliquot of the resuspended urine pellet was subjected to the following pre-treatment. First, a 100-µL aliquot of the polyclonal antibody produced against LipL32 (1:1,000) was added and the mixture incubated for 30 min at 37°C. The mixture was then washed with 1 mL of PBS, centrifuged twice (11,000 × g, 5 min), and finally 0.5 mL (containing 5.0 × 105) of the coated beads was added. Magnetic separation of pathogenic Leptospira from urine samples and the subsequent washing steps were carried out (Invitrogen BeadRetriever system; Life Technologies). Leptospira were selectively concentrated from the 0.5 mL of processed samples obtained from the coated magnetic beads protocol, as described above. The final product of the IMS process was suspended in 0.5 mL of PBS for DNA extraction and purification followed by qPCR, as described below.
DNA extraction and purification
The sample obtained from the IMS step or direct from the re-suspended bacterial pellet was mixed with 500 μL of lysis buffer (2 mM EDTA, 400 mM NaCl, 10 mM Tris-HCl [pH 8.0], and 0.6% SDS) and 5 μL of proteinase K (10 μg/μL; MilliporeSigma) in a 1.5-mL microcentrifuge tube (Eppendorf; MilliporeSigma). The mixture was incubated at 56°C for 1 h, then 500 μL of 100% ethanol was added. The tubes were left standing for 2 min at room temperature before being vortexed for 5 s and centrifuged (11,000 × g, 5 min). The supernatant was discarded, and the pellet was washed once in 200 μL of 70% ethanol by re-suspension and centrifuged as described above. Next, the pellet was re-suspended in 50 μL of sterile distilled water. The tubes were placed in a dry heating block (Eppendorf) at 100°C for 5 min. The solution was centrifuged briefly (16,000 × g, 30 s) to remove any contaminating material. Finally, a 25-μL aliquot of supernatant was placed into a new tube (Eppendorf) to be used as a template for PCR.
qPCR from urine samples
The DNA templates obtained from either the IMS preparation or directly from the re-suspended pellet obtained from the urine were analyzed (LightCycler 2.0; Roche), using a TaqMan probe and targeting the LipL32 gene, which is specific for pathogenic Leptospira species. 16 The amplification mixture for each sample was as follows: 0.7 μM of primers, 0.15 μM of probe, 10 μL of TaqMan universal master mix (Roche), and 5 μL of DNA template, in a total volume of 20 μL. Samples were amplified using the following parameters: initial denaturation at 95°C for 2 min, followed by 40 cycles of denaturation at 95°C for 5 s, and annealing/elongation at 58°C for 30 s. Negative and positive controls to confirm the validity of the reaction were used as well as negative and positive controls of DNA extraction. The Roche system reports crossing point (Cp) values reflecting the number of pathogenic Leptospira in the sample, and a Cp > 40 was regarded as a negative result.
In vitro evaluation of the IMS-qPCR Leptospira detection capacity
The IMS-qPCR detection system was first subjected to an in vitro preliminary evaluation. Five pure cultures of pathogenic Leptospira (L. interrogans serovar Icterohaemorrhagiae strain Icterohaemorrhagiae; L. interrogans serovar Bratislava strain Jez Bratislava; L. interrogans serovar Canicola strain Hond Utrecht IV; L. interrogans serovar Pomona strain Pomona; L. borgpetersenii serovar Tarassovi strain Perepelitsin) were used. From these pure cultures, Leptospira concentration was estimated using direct qPCR according to the genome-equivalent estimation principle described below. Four dilutions with a known concentration of Leptospira (101, 102, 103, and 104 bacteria/mL) were then evaluated by IMS-qPCR using the same quantification method.
Estimation of pathogenic Leptospira shedding level
Pathogenic Leptospira cell numbers (genome equivalents) were estimated according to a published protocol 3 using the molecular weight of the genome of L. interrogans serovar Hardjo type Prajitno strain Hardjo-prajitno (GenBank accession EU357983.1) to establish a standard curve for estimation of Leptospira numbers by Roche 2.0 real-time PCR, according to the following equation:

Statistical analysis
The pathogenic Leptospira detection results obtained by direct PCR and IMS-qPCR were compared using the McNemar test. Bacterial load differences detected by the 2 detection systems were assessed. Because both direct PCR and IMS-qPCR results were not normally distributed, the Wilcoxon matched-pairs signed-rank test was used. For all statistical analyses, p ≤ 0.05 was considered significant. Statistical analyses were done using Prism 6 software (GraphPad).
Results
The SDS-PAGE Coomassie blue stain showed a similar pattern for all 6 pathogenic Leptospira strains (Fig. 1A). The specific immunoreactivity of a major band of the expected molecular size (32 kDa) in all strains was confirmed by western blot (Fig. 1B). Minor bands of different molecular weights were detected, possibly derived from cleavage of the LipL32 target protein. A specific band of low molecular weight (< 9 kDa) was expected for P2 (Fig. 1B). A markedly diminished immunoreaction was observed when antibody was pre-absorbed with P2 immunogenic peptide for all bacterial protein samples (Fig 1C). In the sample containing the P2 immunogenic peptide used to produce antibodies, a total loss of immunoreactivity was observed with the pre-absorbed antibody (Fig. 1C).
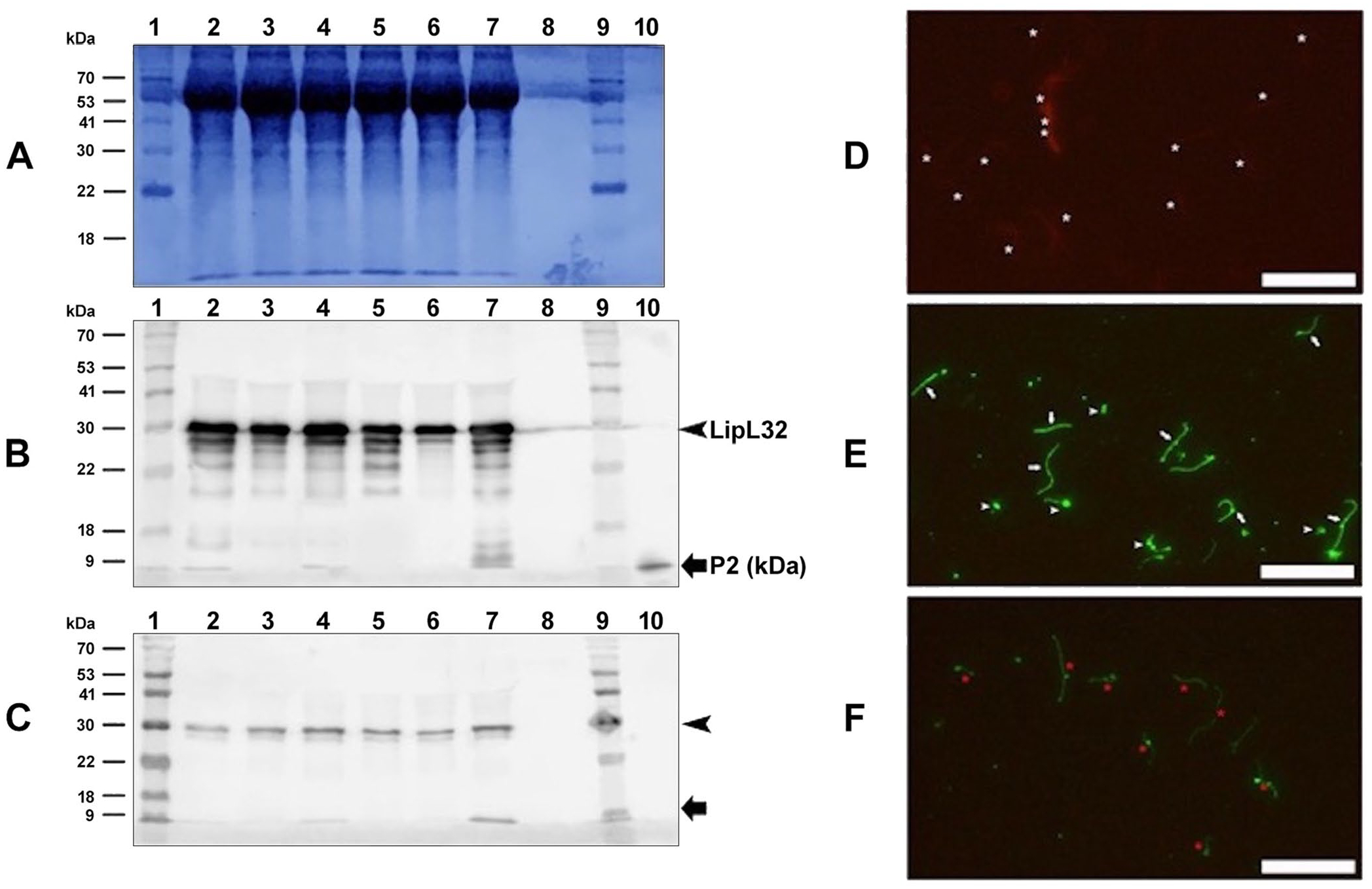
Detection of LipL32 protein in pathogenic Leptospira.
A negative indirect immunofluorescence result was obtained when cell samples were incubated with only secondary antibodies (Fig. 1D). With the same technique, the signal was compatible with the presence of LipL32, with a positive and strong signal for LipL32 in all cells present in the smears, including those cells with a polymorphic size pattern (Fig. 1E). A weak signal was obtained when cell samples were incubated using the anti-Lip32 antibody that had been pre-absorbed with P2 immunogenic peptides (Fig. 1F).
The IMS-qPCR technique with polyclonal antibodies was optimal at 1:1,000 serum dilution (data not shown). In the in vitro evaluation, statistically significant differences were observed from dilutions 101–104 (Fig. 2). The quantitative analysis of IMS-qPCR when used on pure cultures showed that detection of Leptospira loads of < 102 bacteria/mL was not possible.
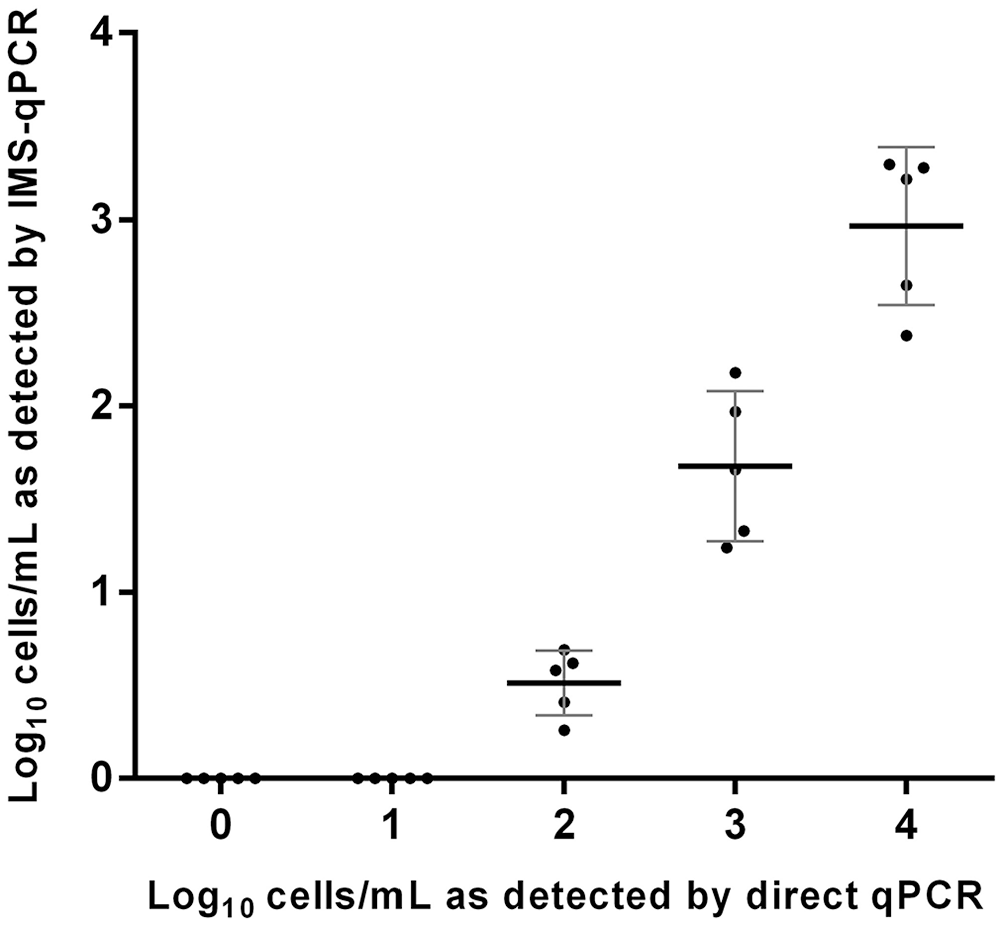
Analytical sensitivity estimation of the immuno-magnetic separation qPCR (IMS-qPCR) compared with direct qPCR for pathogenic Leptospira in different dilutions of pure cultures, expressed in bacteria/mL. Plots represent the mean (central bar) ±1 SD for different culture populations. Statistically significant differences were observed from dilutions 101–104 (p < 0.05).
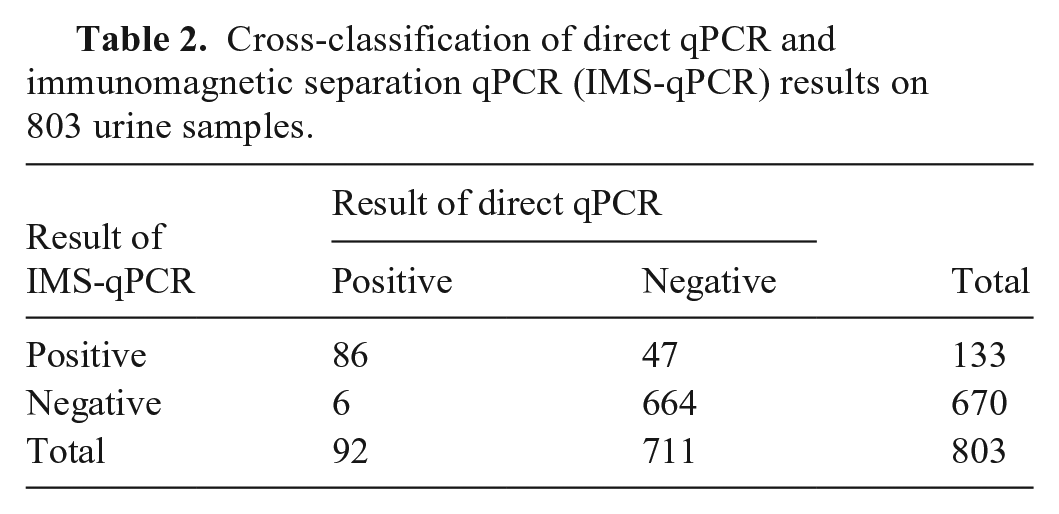
Urine samples from the 38 cattle herds were positive for leptospires by both direct qPCR and IMS-qPCR, with a greater number detected by the latter. Overall, 133 of 803 (16.6%) of the urine samples were IMS-qPCR positive; only 92 of 803 (11.5%) were direct qPCR-positive (p = 0.0001; Table 2). More specifically, 41 samples positive by IMS-qPCR within the range of 100–103 bacteria/mL gave negative results for direct qPCR (Table 3). Statistically significant differences in median load estimation values of the positive samples were observed between the IMS-qPCR assay (4.69 log10 bacteria/mL for the 133 positive urine samples) and the direct qPCR assay (1.49 log10 bacteria/mL for the 92 positive urine samples; Fig. 3).
Cross-classification of direct qPCR and immunomagnetic separation qPCR (IMS-qPCR) results on 803 urine samples.
Frequency distribution of positive direct qPCR results for 133 urine samples with the indicated bacterial load as determined by immunomagnetic separation (IMS) qPCR.

n in parentheses.
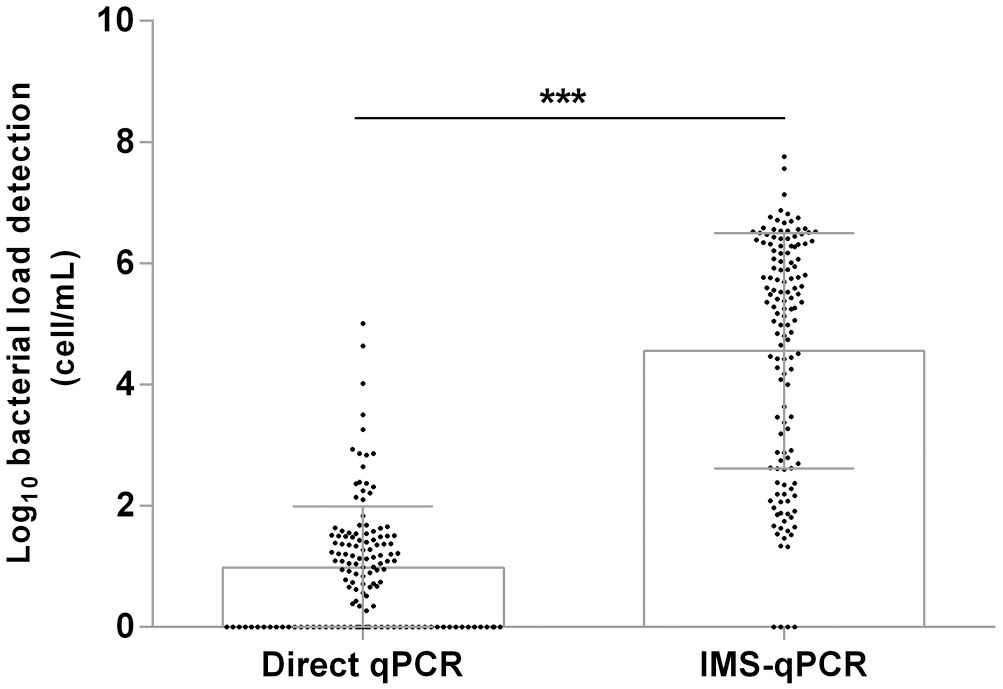
Comparison of the bacterial load detection (cells/mL) for the 133 urine samples positive in the immunomagnetic separation qPCR (IMS-qPCR) assay and the 92 urine samples positive in the direct qPCR assay. *** = p < 0.05 significant difference on paired comparison.
Discussion
Our results confirm specific binding of our anti-LipL32 antibody in the outer membrane region of pathogenic Leptospira, which is consistent with knowledge of the LipL32 protein as an immunogenic target.1,7 This finding was a key element for the implementation of the IMS proposed protocol to be used in urine samples.
The IMS-qPCR technique, when used on pure cultures with known concentrations of pathogenic Leptospira, had limited recovery capacity, resulting in lower sensitivity than qPCR. On the other hand, different results were observed from pathogenic Leptospira obtained from clinical urine samples, especially in bacterial load estimation values of the positive samples. This better performance with clinical samples may be explained by the fact that, for Leptospira strains under laboratory experimental conditions, a significant decrease of LipL32 protein in the outer membrane is expected, 14 possibly because of down-regulation of the LipL32 gene when the pathogen is in a non-infectious environment. In contrast, LipL32 protein is normally expressed in pathogenic Leptospira from infected tissue, and the O-antigen from Leptospira LPS is significantly reduced, leaving LipL32 more exposed. 12 Therefore, the natural Leptospira infectious state may influence the difference in efficiency of the IMS-qPCR protocol. The fact that LipL32 is over-expressed and more exposed on the surface would improve the binding capacity of our anti-LipL32 antibodies and enhance the separation efficiency of IMS when used on clinical samples. The IMS-qPCR technology that we used on clinical samples provided results that yielded a significantly higher number of positive results and with a higher estimated bacterial load (> 3 log10 differences) than direct qPCR in cattle clinical urine samples obtained under field conditions.
There are several possible reasons for the dramatically higher sensitivity of IMS-PCR than direct PCR that we found. Most of these explanations are focused on the effectiveness of an IMS system defined by its antigen-binding capacity or capture efficiency. 18 Furthermore, although direct PCR loses sensitivity because of PCR inhibitors in urine samples, 15 the IMS-qPCR can remove PCR inhibitors during the processing of clinical urine samples. Hence the IMS-qPCR technique improves the analytical sensitivity over traditional techniques. For direct qPCR assays, inhibitors may be removed using specialized DNA extraction kits, but those kits are expensive, making the IMS-qPCR even more advantageous.
The development of the IMS-PCR technique with antibodies against only Leptospira borgpetersenii serovar Hardjo has been reported. 17 An improvement in diagnostic sensitivity in bovine urine samples has been reported for the same serovar. 19 However, in both these reports, the application was limited to one serogroup. Our results show a detection capacity for multiple serogroups and serovars of Leptospira. A previous study 5 produced monoclonal antibodies targeting the LipL32 antigen using a recombinant protein instead of peptides as we did. However, unlike our study, the difference in bacterial concentration detection between PCR and IMS-PCR reported in this prior study was only 1 log10 value. 5
Our novel detection tool had a much higher analytical sensitivity than direct urine qPCR. This high capacity to detect Leptospira-infected animals that shed the pathogen in low concentration will significantly strengthen leptospirosis control efforts in cattle herds.
Footnotes
Acknowledgements
We thank all of the farmers for helping with sample collection.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by Proyecto FIC 15-8 (código BIP 30421496), del Gobierno Regional de Los Ríos, Chile.