
Abstract
Bovine hepacivirus (BoHV) is closely related to the hepatitis C virus (HCV) in humans and can cause both acute and chronic liver infections in cattle. BoHV was first identified in Ghana and Germany in 2015 and since then it has been detected and characterized in other countries around the world, but no strains have been sequenced from U.S. cattle. To date, BoHV has been classified into 2 genotypes (1 and 2), with genotype 1 being further divided into 11 subtypes (A–K). However, the true genetic diversity of BoHV is likely underestimated given limited surveillance and a lack of published genome sequences. Here, we sequenced 2 nearly complete BoHV genomes from serum samples collected in 2019 from beef cattle in Missouri. Sequence comparisons and phylogenetic analysis showed that isolate MARC/2019/60 had high sequence homology with genotype 1, subtype E isolates from China. In contrast, isolate MARC/2019/50 represented a novel BoHV subtype within genotype 2. Thus, we report the first genomic characterization of BoHV isolates from U.S. cattle, and the second complete BoHV2 genome worldwide. This work increases our knowledge of the global genetic diversity of BoHV and demonstrates the co-circulation of divergent BoHV strains in U.S. cattle.
Hepatitis C virus (HCV; Hepacivirus hominis) is a major human pathogen that can cause both acute and chronic liver infections. Chronic HCV infection can lead to liver damage, including inflammation, cirrhosis, and cancer. Until recently, HCV was the only species in the Hepacivirus genus within the family Flaviviridae. However, the combination of improved sequencing methods and advanced computational tools has led to the discovery of many novel HCV-like viruses in diverse animal species, including a variety of mammalian and non-mammalian hosts.10,12,17,18
Bovine hepacivirus (BoHV; Hepacivirus bovis, formerly hepacivirus N) was first identified in 2015 in cattle from Ghana and Germany.2,6 BoHV has subsequently been detected in bovine serum samples from Brazil,5,7 the United States, 15 Turkey, 21 Italy, 9 China, 13 Bulgaria, 4 and Uganda. 3 Like HCV, BoHV is hepatotropic and can cause both acute and persistent liver infections in cattle. 1 Although BoHV infection has not been associated with overt liver damage or clinical disease, much remains unknown about the pathogenesis of BoHV infections in cattle. Furthermore, the recent detection of BoHV in wild boars in Italy 8 and a red deer from the Czech Republic 4 raise further questions about the host range and zoonotic potential of BoHV.
BoHV has a single-stranded RNA genome that is ~9 kb long and encodes a single polyprotein that is cleaved by host and viral proteases into 3 structural proteins (C, E1, E2) and 7 nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, NS5B). BoHV genotyping and subtyping is performed according to HCV classification criteria in which a novel genotype is defined by having <77% amino acid identity compared to all known strains, and a novel subtype is defined by having <85% nucleotide identity compared to sequences of the same genotype.4,17 To date, BoHV has been classified into 2 genotypes (1 and 2), with genotype 1 being further divided into 11 potential subtypes (A–K).4,16 However, the true distribution and genetic diversity of BoHV is likely underestimated given limited surveillance and few published genome sequences.
Here, we sequenced 2 nearly complete BoHV genomes from serum samples collected from 2 beef cattle in Missouri, USA. In June 2019, we collected whole blood from 39 beef cattle persistently infected (PI) with bovine viral diarrhea virus (BVDV; Pestivirus bovis), a related virus in the family Flaviviridae, genus Pestivirus. These samples were collected as part of an epidemiologic study of BVDV in U.S. beef cattle under a University of Nebraska–Lincoln Institutional Animal Care and Use Committee approved protocol (IACUC protocol 1915). These cattle were privately owned by a commercial stocker operation and were originally purchased from livestock auctions in Missouri. Upon purchase, the operation tested all cattle <272 kg (600 lb) for BVDV infection using a noncommercial antigen ELISA. The cattle that tested positive for BVDV were held in pens segregated from other cattle and retested for BVDV infection a minimum of 2 wk after the initial positive test to determine whether the cattle were transiently infected or PI with the virus. We collected blood samples from BVDV PI cattle via jugular venipuncture (Suppl. Table 1). We separated serum by centrifugation at 1,600 × g for 15 min at 4°C and stored the serum at −80°C in 1-mL aliquots until use.
We genotyped the BVDV strains in the 39 serum samples by Sanger sequencing a 283-bp fragment of the 5′-untranslated region (5′-UTR) as described previously. 19 We then selected 24 representative samples for complete genome sequencing. Prior to sequencing, we nuclease-treated the serum samples to enrich encapsidated viral RNA and degrade unprotected host and environmental nucleic acids. 20 The remaining RNA was isolated using a phenol and guanidine isothiocyanate reagent (TRIzol LS; Life Technologies) and used to prepare indexed sequencing libraries (TruSeq Stranded mRNA kit; Illumina), according to the manufacturer’s instructions, without the initial step of poly(A) selection. 19 We sequenced libraries with a massively parallel sequencing machine with high-output kits (NextSeq 2000; Illumina) to generate 2.6–81 million reads per sample library (Suppl. Table 1). We processed and assembled raw sequencing reads using a commercial DNA analysis software package (Geneious Prime, v.2022.2.2; Dotmatics). Index adapters and low-quality reads were removed from raw sequence reads using BBDuk (v.38.84; Dotmatics), and the trimmed reads were de novo assembled using the SPAdes assembler for metagenomic datasets (v.3.15.2, https://cab.spbu.ru/software/spades/). We then compared the assembled contigs >500 nucleotides (nt) to known sequences in the National Center for Biotechnology Information (NCBI) nucleotide database using MegaBLAST as implemented in Geneious Prime.
In addition to the expected BVDV genome sequence, we assembled a complete BoHV genome from one serum sample (serum 2019/60; Suppl. Table 1). The assembled contig was 8,831 nt long and had 88% nt sequence identity to GenBank MG257793, a BoHV1 isolate sequenced from a bovine serum sample collected in 2017 in China. To determine the number of hepacivirus reads in the dataset, we mapped the sequencing library to the de novo assembled BoHV sequence. A total of 295,654 reads out of 5,197,836 (5.7%) mapped to the contig at a mean depth of 2,092×. As comparison, 54,649 reads (1.1%) mapped to the de novo assembled BVDV contig at a mean depth of 62×. We submitted the sequence for this new BoHV isolate (MARC/2019/60) to GenBank under accession OR543978.
We also assembled a partial BoHV genome sequence from a second serum sample (serum 2019/50; Suppl. Table 1). The contig was 651 nt and had 84.7% nt sequence identity to GenBank ON375567, a partial BoHV2 sequence (835 nt) from a red deer serum sample from the Czech Republic. 4 We next mapped the sequencing library from serum sample 2019/50 to GenBank MN691105, the only publicly available complete BoHV2 genome, to identify any additional hepacivirus reads in the dataset. A total of 5,331 reads out of 22,171,224 (0.024%) mapped to MN691105, covering 51% of the reference genome sequence at a mean depth of 46×. As comparison, 3,349,232 reads (15%) mapped to the assembled BVDV contig at a mean depth of 20,726×.
To fill gaps between sequence information obtained from short-read sequencing, we constructed long-read sequencing libraries according to the manufacturer’s instructions (PacBio SMRTbell prep kit 3.0; Pacific Biosciences) with one important modification. To ensure amplification of subgenomic RNA and viral RNA genomes lacking a poly(A) tail, we first added poly(A) tails to the 3′-end of the RNA using a poly(A) tailing kit (New England Biolabs). We then sequenced libraries with single-molecule, real-time sequencing technology (Sequel II system; Pacific Biosciences) generating ~2.4 million reads with a quality score >Q30. Reads were error-corrected and trimmed using the SMRT Analysis server according to the IsoSeq analysis pipeline (https://www.pacb.com/products-and-services/analytical-software/smrt-analysis/). This pipeline identifies and removes poly(A) tails and primer sequences, identifies read strandedness, and removes artificial concatemers. We analyzed the full-length non-concatemer (FLNC) reads in Geneious by mapping them to the BoHV2 reference genome MN691105 to discriminate hepaciviral reads from others in the dataset. Of the 2,353,654 HiFi long-reads, 1,218 reads (0.05%) mapped to MN691105, covering 98% of the reference genome, with only a portion of the 5′-UTR missing. The lengths of the mapped reads ranged from 378 nt to 6,771 nt with a mean read length of 1,836 nt. The mapped hepacivirus reads were then de novo assembled together with the Illumina short-reads using the SPAdes assembler to produce a contig that was 8,629 nt long and had 82% nt sequence identity to GenBank MN691105. We submitted the sequence for this isolate (MARC/2019/50) as GenBank OR566925.
To explore the evolutionary relationships among BoHV, we analyzed our 2 novel hepacivirus genomes along with all publicly available complete BoHV genomes (n = 34; NCBI database accessed 2023.07.28). We also included the partial BoHV2 sequence reported from Brazil (MK695669; 6,102 nt). We aligned the open-reading frame (ORF) encoding the polyprotein with MUSCLE (v.3.8.425; https://www.drive5.com/muscle/) and constructed a maximum-likelihood tree in MEGAX (v.10.2.4; https://www.megasoftware.net/) employing the general time reversible (GTR) nucleotide substitution model with gamma (G) rate heterogeneity and invariant sites (I; i.e., GTR+G+I model). The support values on the tree were calculated from 500 bootstrap replicates (Fig. 1A). In addition, we produced a neighbor net phylogenetic network in SplitsTree (CE v.6.1.13 11 ; Fig. 1B) and a matrix of pairwise identities from aligned nucleotide and amino acid sequences (Suppl. Tables 2, 3, respectively).
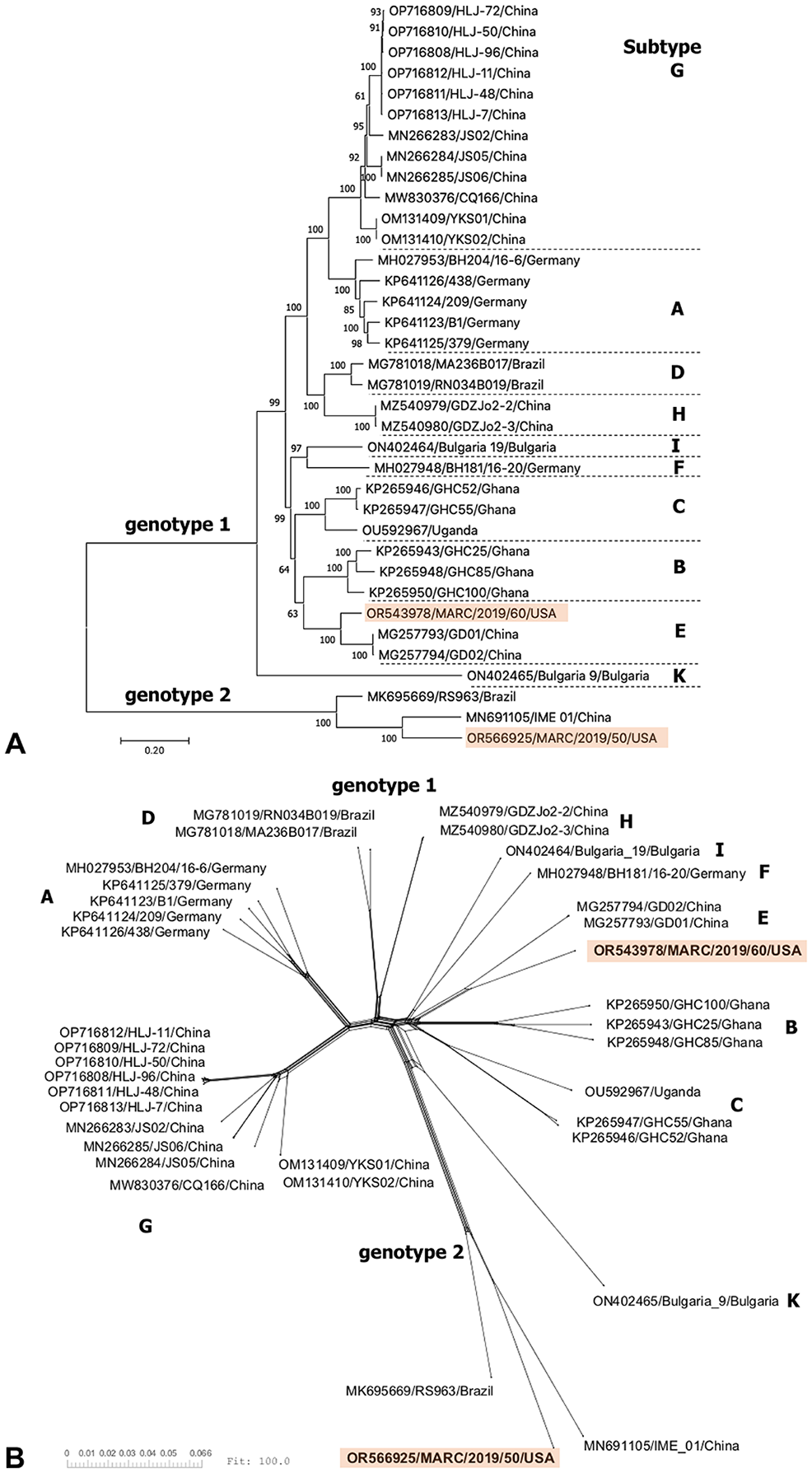
Phylogenetic analysis of bovine hepacivirus polyprotein coding sequences.
According to phylogenetic relationships and sequence distances, the new U.S. BoHV1 isolate MARC/2019/60 (OR543978) is classified as genotype 1, subtype E along with BoHV1 isolates GD01 (MG257793) and GD02 (MG257794) from China (Fig. 1). 13 MARC/2019/60 shared 88.2% nt sequence identity with both isolates (Suppl. Table 2). Six other BoHV1 NS3 gene sequences from Turkey were previously shown to cluster with the subtype E variants from China. 4 Thus, to date, BoHV1 subtype E has been reported in China, Turkey, and the United States.
The U.S. isolate MARC/2019/50 (OR566925) is classified as genotype 2 along with the BoHV2 isolate IME_01 (MN691105) from China and RS963 (MK695669) from Brazil (Fig. 1).5,14 MARC/2019/50 shared 82.5% nt identity with IME_01 (MN691105) and 80.5% nt identity with RS963 (MK695669), suggesting that MARC/2019/50 represents a novel subtype within genotype 2 (Suppl. Table 2). Although it is common to see sequences clustering by their geographic origin, additional surveillance is needed to better understand the global geographic distribution of BoHV genotypes and subtypes. In particular, further epidemiologic surveys should be completed to better define the prevalence and diversity of BoHV strains in the United States.
We demonstrate here the co-circulation of genotype 1 and 2 BoHV strains at a single commercial stocker operation in Missouri, USA. Although the transmission routes for BoHV have not been fully characterized, contact with blood from infected individuals is likely the primary route (e.g., from contaminated vaccine needles or possible mechanical transfer by ticks 22 ). Vertical transmission from the dam to fetus has also been hypothesized, although additional research is needed. It is of note that the isolates that we characterized were detected in BVDV PI cattle. In PI animals, BVDV is present in all tissues, including the liver. How concurrent BVDV and BoHV liver infections may impact clinical disease progression or viral pathogenesis is unknown.
Supplemental Material
sj-pdf-1-vdi-10.1177_10406387231225656 – Supplemental material for Two bovine hepacivirus genome sequences from U.S. cattle
Supplemental material, sj-pdf-1-vdi-10.1177_10406387231225656 for Two bovine hepacivirus genome sequences from U.S. cattle by Aspen M. Workman, Gregory P. Harhay, John T. Groves and Brian L. Vander Ley in Journal of Veterinary Diagnostic Investigation
Supplemental Material
sj-xlsx-2-vdi-10.1177_10406387231225656 – Supplemental material for Two bovine hepacivirus genome sequences from U.S. cattle
Supplemental material, sj-xlsx-2-vdi-10.1177_10406387231225656 for Two bovine hepacivirus genome sequences from U.S. cattle by Aspen M. Workman, Gregory P. Harhay, John T. Groves and Brian L. Vander Ley in Journal of Veterinary Diagnostic Investigation
Supplemental Material
sj-xlsx-3-vdi-10.1177_10406387231225656 – Supplemental material for Two bovine hepacivirus genome sequences from U.S. cattle
Supplemental material, sj-xlsx-3-vdi-10.1177_10406387231225656 for Two bovine hepacivirus genome sequences from U.S. cattle by Aspen M. Workman, Gregory P. Harhay, John T. Groves and Brian L. Vander Ley in Journal of Veterinary Diagnostic Investigation
Footnotes
Acknowledgements
We thank Susan Hauver, Jaden Carlson, and the U.S. Meat Animal Research Center Core Facility for technical support, and Janel Nierman for secretarial support. The use of product and company names is necessary to accurately report the methods and results; however, the USDA neither guarantees nor warrants the standard of the products. The use of names by the USDA implies no approval of the product to the exclusion of others that may also be suitable. The USDA is an equal opportunity provider and employer.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Funding for this research was provided by the USDA–ARS appropriated project 3040-32000-036-00D.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.