
Abstract
Virus was detected in the central nervous system (CNS) tissue of 11 horses from Victoria that died displaying neurological symptoms during an outbreak of disease in Australia in 2011. Five horses were identified as being infected with Murray Valley encephalitis virus (MVEV) and 6 as being infected with West Nile virus subtype Kunjin (WNVKUN). Analysis of partial sequence information from the NS5 and E genes indicated that the MVEVs within the samples were highly homogenous and all belonged to lineage I, which is enzootic to the tropical regions of northern Australia. Likewise, analysis of partial NS5 and E gene and full genome sequences indicated that the WNVKUN within the samples were also highly homogenous and clustered with WNV lineage 1, clade b, which is consistent with other WNVKUN isolates. Full genomes of 1 MVEV isolate and 2 WNVKUN isolates were sequenced and characterized. The genome sequences of Victorian WNVKUN are almost identical (3 amino acid differences) to that of the recently sequenced WNV isolate WNVNSW2011. Metagenome sequencing directly from CNS tissue identified the presence of WNVKUN and MVEV within infected CNS tissue.
Introduction
Murray Valley encephalitis virus (MVEV) and West Nile virus subtype Kunjin (WNVKUN) are closely related viruses that belong to the Japanese encephalitis group within the genus Flavivirus. 8 MVEV and WNVKUN are both zoonotic viruses endemic to Australasia. Although uncommon, MVEV poses a significant human health risk causing symptoms ranging from mild febrile illness to potentially fatal encephalitis. 7 WNV is a widely distributed flavivirus that has been associated with fatal meningoencephalitis.30,40 WNVKUN is the only WNV variant identified in Australia; the variant can also cause febrile illness and encephalitis but cases of disease caused by WNVKUN in human beings or horses are rare. In human beings, the symptoms are milder than those caused by MVEV and WNV.15,17,33
MVEV and WNVKUN viruses have transmission cycles that involve water birds as the vertebrate host 6 and mosquitoes as the vector. 36 The primary vector of these viruses in Australia is Culex annulirostris (common banded mosquito), which is widely distributed across Australia, particularly in freshwater environments. 36 MVEV is predominantly found in northern Australia and Papua New Guinea but has also been detected farther south when environmental conditions are conducive. 22 The distribution of WNVKUN is broader than MVEV; WNVKUN is enzootic across tropical northern Queensland, the Northern Territory, and Western Australia, and is regularly detected in southeastern Australia. 32
There is considerable anecdotal evidence that, in years where MVEV outbreaks have occurred in human beings, there is also an increased incidence of neurological disease in horses. 38 Sera collected from horses in 1951, the year of a significant outbreak of MVEV in human beings in Victoria, had antibodies to MVEV. 29 In 1974, a year that saw a widespread outbreak of MVEV in human beings in New South Wales (NSW) and Victoria, there were many reports of neurological disease in horses in NSW, and sera from horses in NSW also tested positive for antibodies for MVEV and Ross River virus. 13 Symptoms of MVEV and WNVKUN in horses are similar, with the animal exhibiting changes in temperament, in-coordination, weakness, muscle paralysis, and tremors (Australian Government, Department of Agriculture, Fisheries and Forestry: 2011, Disease in horses due to insect borne viruses. Available at: http://www.outbreak.gov.au/pests_diseases/pests_diseases_animals/disease-in-horses-insect/docs/horse-insect-200511.pdf). Although both MVEV and WNVKUN can cause disease symptoms in animals, including human beings and horses, animals are considered to be dead-end hosts for these viruses.23,27
In February 2011, an increased number of horses showing muscle and joint soreness and signs of neurological disease were reported in the southern states of Australia (Queensland Government, Department of Agriculture, Fisheries and Forestry: 2011, Neurological disease in horses. Available at: http://www.daff.qld.gov.au/4790_20274.htm). In Victoria, diagnosis of many of these cases indicated the causal agent was likely to be attributed to an arbovirus infection caused by 1 or more of the 3 arboviruses circulating at the time; the alphavirus Ross River virus and the flaviviruses MVEV and WNV. However, only the flaviviruses were thought to be causing the neurological cases (Queensland Government, Department of Agriculture, Fisheries and Forestry: 2011). The current study describes the identification of 2 viral agents, MVEV and WNVKUN, in tissue from the central nervous system (CNS) of deceased horses and the phylogenetic analysis of sequence data from the MVEV and WNVKUN strains circulating during the outbreak. In addition, genome sequence analysis of propagated viral isolates from the 2011 disease outbreak, and metagenome sequence analysis directly from CNS tissue are also reported.
Materials and methods
Virus isolation
CNS tissue was homogenized and resuspended in viral transport medium (VTM; 10% w/v), which is a 3.7% (w/v) brain-heart infusion broth containing 3 μg/ml of benzyl penicillin, 5 mg/ml of streptomycin, 100 μg/ml of gentamicin, and 1 μg/ml of amphotericin B, and inoculated onto nearly confluent C6/36 cell line derived from Aedes albopictus (Asian tiger mosquito). Following adsorption of the inoculum for 1 hr at 32°C, maintenance medium was added, and the cultures were incubated at 32°C with a 5% CO2 atmosphere for 5 days before the cells were subjected to 2 freeze–thaw cycles to release the virus. The supernatant was passaged onto nearly confluent cell lines BHK (baby hamster kidney)-21 and SVP (derived from the pig kidney cell line) 9 and incubated at 37°C with a 5% CO2 atmosphere for up to 7 days. When a cytopathic effect was detected, the cell cultures were frozen and thawed twice, and the supernatant was harvested. The supernatant was clarified by centrifugation at 1,000 × g for 5 min and was stored at −80°C until tested
Polymerase chain reaction and amplicon sequencing
RNA was extracted from CNS tissue samples in VTM and from supernatant harvested from propagated virus using a commercial system a in accordance with the manufacturer’s instructions. Purified nucleic acid was stored at −20°C if not used immediately. MVEV- and WNV-specific reverse transcription quantitative polymerase chain reaction (RT-qPCR) was performed as previously described11,35 using a commercial kit. b
Pan-flavivirus conventional nested PCR primers targeting the NS5 gene 14 (with additional M13 tails on the internal primers to facilitate sequencing of the amplicons) were used to generate amplicons for sequencing and phylogenetic analysis as well as to confirm RT-qPCR results. The first-round RT-PCR was performed using a commercial one-step RT-PCR kit c in a 25-μl reaction containing 1 × commercial reaction buffer, 200 nM of each primer, 0.5 μl of Taq enzyme mix, and 5 μl of RNA template under the following cycling conditions: 50°C for 45 min, 94°C for 2 min followed by 4 cycles of 94°C for 20 sec, 45°C for 1 min, and 72°C for 1 min, followed by 46 cycles of 94°C for 20 sec, 50°C for 30 sec, and 72°C for 1 min and ending with 72°C for 5 min. The second-round PCR was performed using a commercial PCR kit d in a 25-μl reaction containing 1 × reaction buffer, 1.5 mM of MgCl2, 200 nM of each primer, 200 μM of each deoxyribonucleotide triphosphate, 1 U Taq enzyme, and 2.5 μl of the first-round product as the template. Cycling conditions for the second round were 94°C for 2 min followed by 35 cycles of 94°C for 20 sec, 50°C for 30 sec, and 72°C for 30 sec, with a final extension of 5 min at 72°C.
A region of the envelope (E) gene was amplified from MVEV-positive samples using previously described primers. 22 The PCR was performed as previously described, 22 except that a commercial one-step RT-PCR kit c was used in a 25-μl reaction containing 1 × commercial reaction buffer, 200 nM of each primer, 0.5 μl of Taq enzyme mix, and 5 μl of RNA template. The PCR amplicons were separated and visualized on 1.5% agarose gels containing a dye. e Amplification products were purified using a commercial PCR purification kit f and submitted to a sequencing facility g for capillary sequencing. The NS5 amplicons from the nested PCR were sequenced using M13 primers, while the E gene products were sequenced using the amplification primers. Sequence data were compared to the nonredundant database at the National Center for Biotechnology Information (NCBI) using the BLASTn algorithm. 1
Template preparation for sequencing
Cell culture propagated virus (35 ml) and homogenized CNS samples in VTM (0.5 ml) were vortexed thoroughly, centrifuged for 3 min at 10,000 × g, and the supernatant filtered through a 0.45-μm filter to remove any large debris. The filtered supernatant was then ultracentrifuged at 22, 000 × g for 2 hr at 8°C. The supernatant was removed, and the pellet was resuspended in 100 μl of molecular biology grade water. The resuspended pellets were then treated with 7 U of DNase h and 20 U of RNase 1 i to reduce the concentration of nonviral DNA and RNA while preserving entire viral particles. RNA was extracted using a commercial kit j in accordance with the manufacturer’s instructions, and a ribonuclease inhibitor i was used to stabilize the RNA.
Library preparation and sequencing
Construction of 5 libraries from extracted RNA template was conducted following the manufacturers protocol k with minor variation. Briefly, heat fragmentation (94°C for 5 min) was used to sheer RNA before being used in first-strand complementary DNA (cDNA) synthesis, second-strand cDNA synthesis, end repair, A-tailing, and adapter ligation. Initial enrichment PCR (with indexed primers for individual libraries) was performed using a high fidelity DNA polymerase l with the following cycling conditions: initial denaturation for 30 sec at 98°C, followed by 30 cycles of denaturation for 10 sec at 98°C, annealing for 20 sec at 65°C, and extension for 1 min at 75°C to determine the optimal enrichment PCR cycle number for each library (to avoid skewing the representation of the library). Enrichment PCR was then repeated using the optimal cycle number for each library with index primers, quantified using a commercial quantification kit, m and run on a commercial system n to determine size distribution and purity. As an additional quality control, specific RT-qPCR for WNV 11 and MVEV 35 were used to confirm the presence of the target organism in the enriched libraries. Libraries were then pooled, quantified using a commercial kit, m and quality checked on a commercial system. n Clustering was performed on a commercial cluster generation system o according to the manufacturer’s instructions and then sequenced on a commercial sequencer. p
Paired-end sequence reads were mapped to reference genomes (MVE-1-51 and WNV K6453) using the Burrows–Wheeler Alignment Tool 25 and Samtools 26 was used to convert, sort, index, and write consensus files from the aligned data. Paired end reads from the CNS samples were also mapped to a database of all viruses at NCBI from vertebrate hosts (http://www.ncbi.nlm.nih.gov/genomes/GenomesGroup.cgi?opt=virus&taxid=10239&host=vertebrates) using the same method. Velvet 41 (version 1.1.06) was used for de novo genome assembly of the paired end data using the VelvetOptimiser script (version 2.1.4). q The contigs generated were compared to the mapped consensus genomes and the nonredundant database at NCBI using the BLASTn algorithm. 1
Phylogenetic analysis
Partial E gene sequences and full genome sequences of members of the Japanese encephalitis group of the Flavivirus genus (Supplementary Table 1) were aligned individually using MUSCLE. 10 Phylogenetic analysis was performed using the program MEGA5 39 employing the maximum parsimony method; a bootstrap consensus tree was also inferred using 1,000 resamplings of the data. The close-neighbor-interchange algorithm 31 was used to obtain the initial trees with the random addition of 10 replicate sequences.
Results
Virus isolation, PCR, and amplicon sequencing
All 11 CNS tissue samples tested positive for the presence of flaviviruses using the pan-flavivirus RT-nested PCR (Table 1). In addition, out of the 11 samples tested, 5 were positive for MVEV and 6 for WNVKUN using RT-qPCR and partial NS5 sequencing (Table 1). From the 11 CNS tissue samples, 4 flavivirus isolates were successfully propagated (Table 1), and isolates were identified as WNV (WNVKUNV11-02, WNVKUNV11-03 and WNVKUNV11-07) or MVEV (MVEV- V11-10) by PCR amplification of the NS5 gene and sequence analysis.
Virus isolation, reverse transcription quantitative polymerase chain reaction (RT-qPCR) for Murray Valley encephalitis virus (MVEV) and West Nile virus (WNV), and NS5 gene sequencing results for central nervous system tissue samples from 11 horses that died exhibiting neurological disease.*
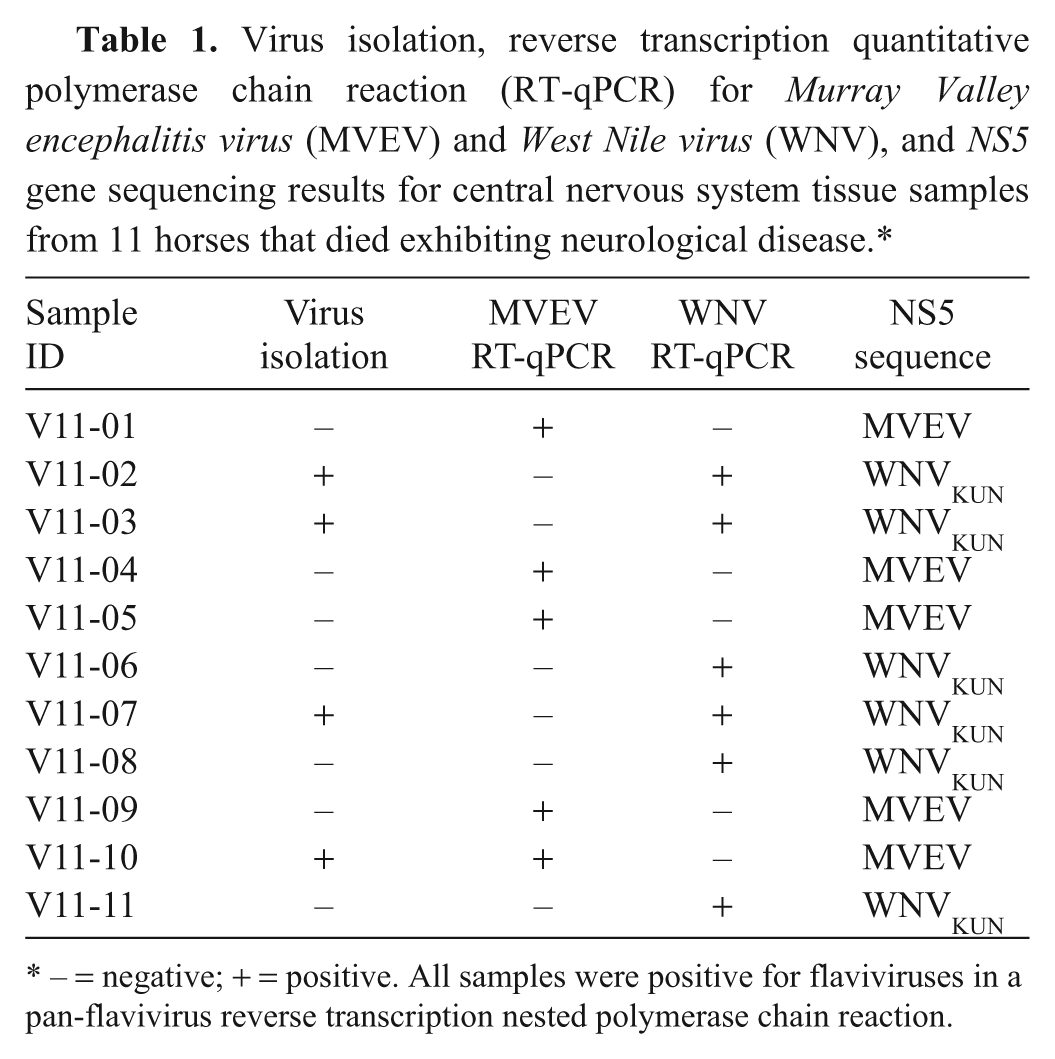
− = negative; + = positive. All samples were positive for flaviviruses in a pan-flavivirus reverse transcription nested polymerase chain reaction.
Partial NS5 sequences from the MVEV-positive samples shared high nucleotide identity (>99%). When compared to sequences in the nonredundant database at NCBI, the NS5 partial sequence of MVEV shared highest nucleotide identity to MVE-1-51 (>97%).
Partial E gene sequence data from MVEV-positive samples shared 100% nucleotide identity, clustered within lineage I of MVEV 22 in phylogenetic analysis, and were identical to 2 MVEV isolates collected in NSW in 2008 (Fig. 1). Within lineage I there is no evidence of clustering based on geographic distribution but there is bootstrap support (74%) for clustering of Australian MVEV isolated in the more recent past (1997 onward). The partial E gene sequence data from Victorian 2011 MVEV samples shared >97% nucleotide identity with MVE-1-51 isolated in Victoria in 1951.
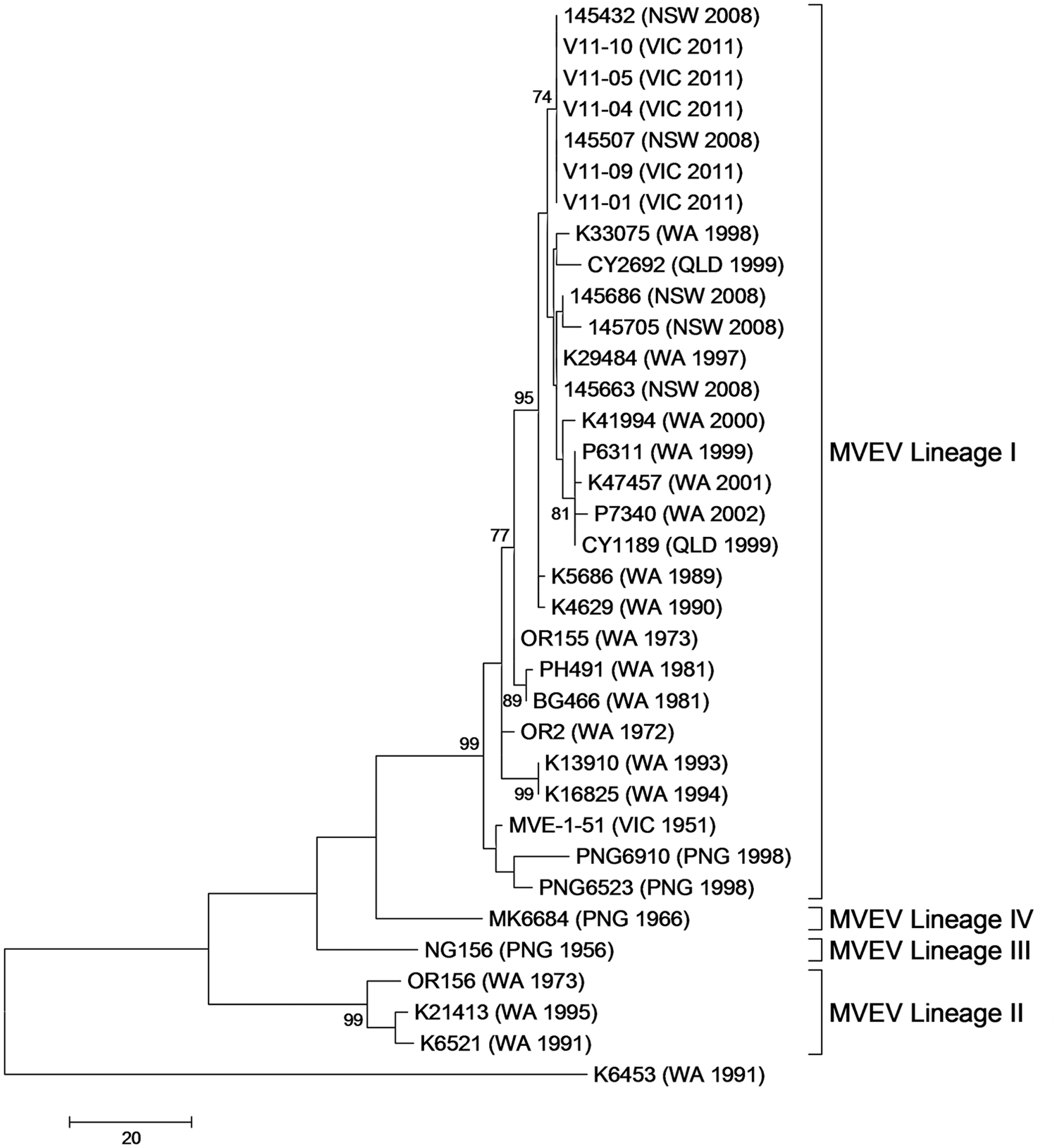
Maximum parsimony tree based on the alignment of a partial region on the envelope (E) gene of Murray Valley encephalitis virus (MVEV). All of the Victorian 2011 MVEV samples cluster with MVEV lineage I.
The partial NS5 region sequenced from 6 WNVKUN-positive samples in the current study share a high degree of nucleotide identity (>99%) with each other and with WNV isolate WNVNSW2011. When compared with sequences in the nonredundant database at NCBI, the Victorian 2011 WNVKUN partial NS5 sequences shared highest nucleotide identity with the WNVKUN strain K6543 (97%), followed by Mitchell River isolates MRM16 (94%; Queensland 1960) and MRM61C (93%; Queensland 1960).
Sequencing
Complete genomes of the virus isolates WNVKUNV11-03, WNVKUNV11-07, and MVEV-V11-10 (GenBank accession nos. JX123030–JX123032) were assembled (> 100 × coverage), and the polyproteins were identified. Whole genome phylogenetic analysis of MVEV-V11-10 showed that this isolate clusters with the MVE-1-51 (Victoria 1951) isolate genome sequence (Fig. 2). MVE-1-51 is also the closest match to MVEV-V11-10 in the nonredundant database at NCBI, with 96% nucleotide identity. Variations in the polyproteins of these isolates are shown in Figure 3.
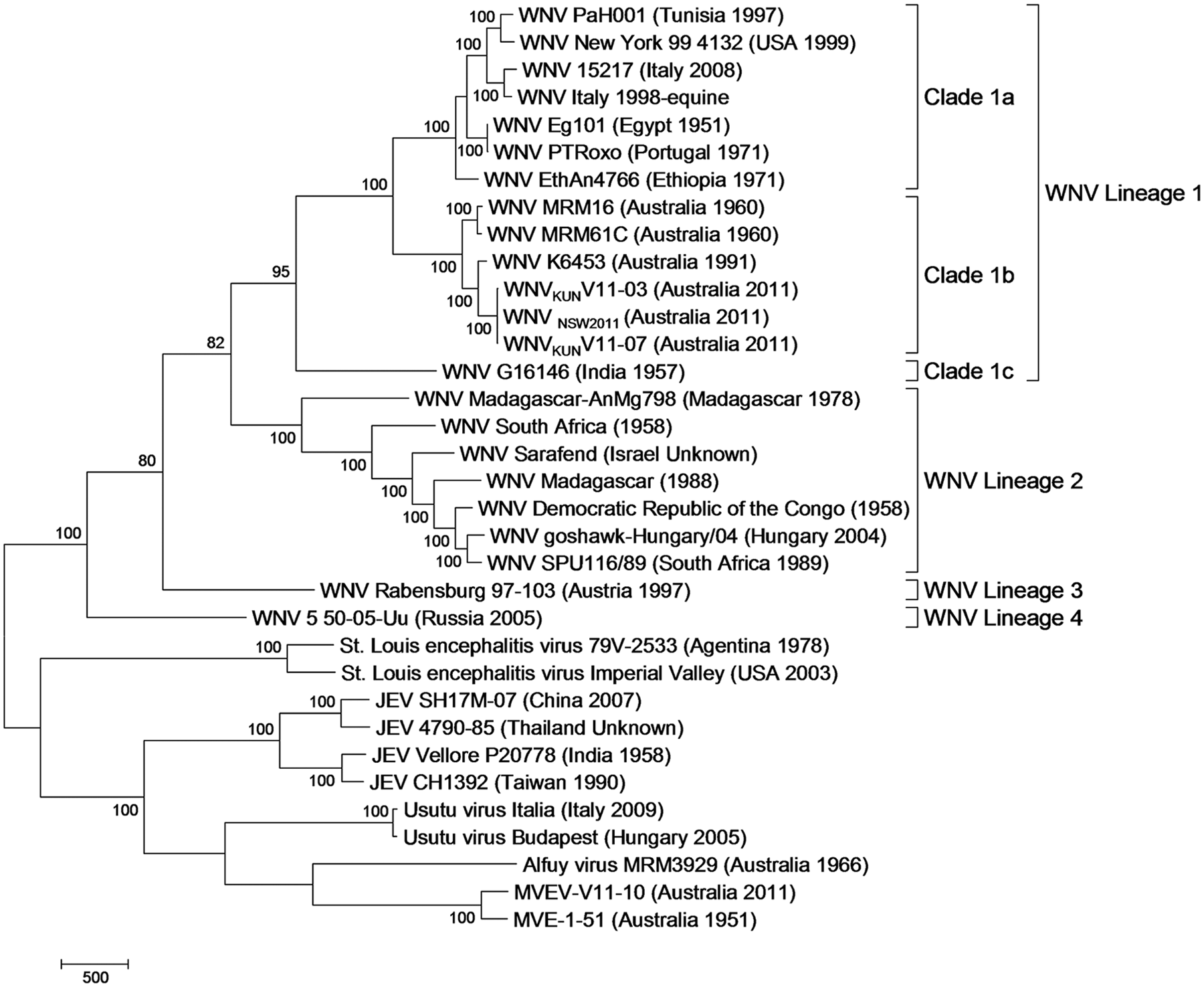
Maximum parsimony tree based on the alignment of complete genomes of viruses in the Japanese encephalitis virus (JEV) group of the Flavivirus. Victorian 2011 West Nile virus (WNV) subtype Kunjin isolates WNVKUNV11-03 and WNVKUNV11-07 cluster with WNV lineage 1, clade b and the Victorian 2011 Murray Valley encephalitis virus (MVEV) isolate MVEV-V11-10 clusters with MVE-1-51.

Amino acid variation across polyproteins of isolates of (
The isolates WNVKUNV11-03 and WNVKUNV11-07 are nearly identical, sharing > 99% nucleotide identity. Both isolates cluster with other characterized WNVKUN isolates including WNVNSW2011, MRM16, MRM61C, and K6453 (Western Australia 1991) in phylogenetic analysis (Fig. 2). Isolates WNVKUNV11-03 and WNVKUNV11-07 are nearly identical to WNVNSW2011 (99% nucleotide identity), and all three 2011 isolates are more similar to K6453 (98% nucleotide identity) than MRM16 and MRM61C (96% nucleotide identity). Variation in the polyproteins of WNVKUNV11-03 and WNVKUNV11-07 compared to other WNVKUN and WNV isolates are shown in Figure 3. There are 3 amino acid differences between 2011 isolates, and K6453 differs from WNVKUNV11-03 and WNVKUNV11-07, and WNVNSW2011 by 23, 22, and 21 amino acids, respectively. There are 52 amino acid differences between the 2011 isolates and Mitchell River isolates.
Within the polyprotein, there are 3 loci where the 2011 WNVKUN has substitutions identical to virulent WNV, which contrasts with 3 other WNVKUN (Fig. 3). The substitutions include a substitution within the nonstructural protein NS5 at residue 49 (2577; I→V) located in the methyl transferase domain of NS5, which is involved in RNA capping and is a binding site for the monoclonal antibody 5H1. 18 There are 6 loci where the 2011 isolates and K6543 have substitutions (as compared with the Mitchell River isolates) identical to virulent WNV (Fig. 3). The substitutions include one of the glycosylation tripeptide (N-Y-S) residues, E154 (polyprotein 446; Fig. 3), which is associated with virulence in most WNV strains, 4 and 3 substitutions in NS2A, a nonstructural protein that has been associated with variation in virulence between WNV and WNVKUN. 2
Phylogenetic analysis was also conducted on a partial region of the E gene (which differs from the region amplified for MVEV) from the genomes of WNVKUNV11-03 and WNVKUNV11-07 to enable comparison to sequence data from other strains of WNVKUN from previous research.22,37 Analysis of the E gene sequence data clustered isolates WNVKUNV11-03 and WNVKUNV11-07 with other WNVKUN isolates (clade 1b) with high bootstrap support (>96%; Fig. 4). Within clade 1b of the E gene phylogenetic analysis, the more recently isolated WNVKUN (1991 onward) clustered together (bootstrap support of 79%).
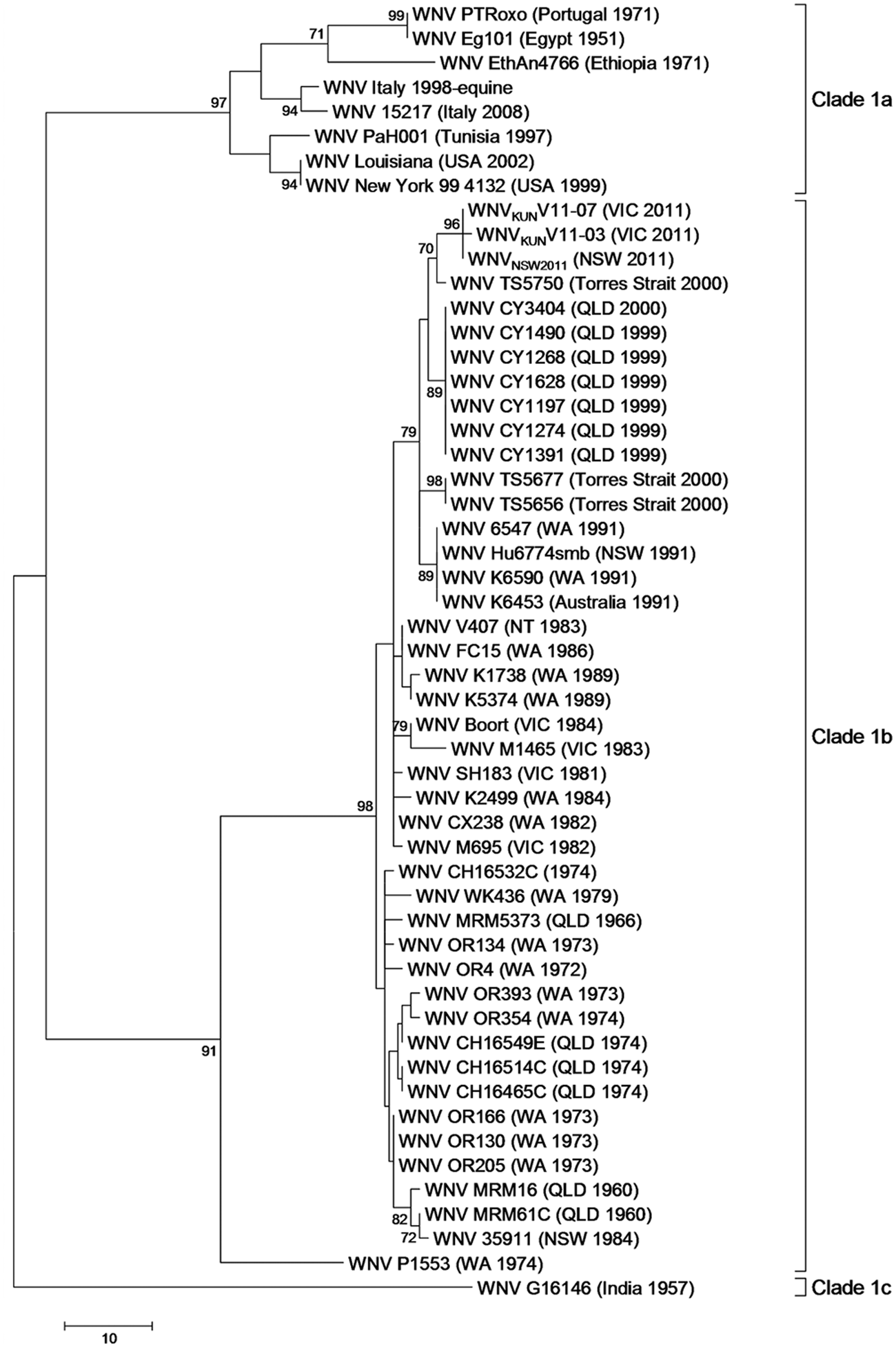
Maximum parsimony tree of West Nile virus lineage 1 based on the alignment of a partial region of the envelope (E) gene.
Sequencing of the CNS metagenome from samples V11-07 and V11-10 successfully identified genome sequence data identical to WNVKUN propagated virus isolate WNVKUNV11-07 and MVEV propagated virus isolate MVEV-V11-10, respectively. The complete genome of WNVKUNV11-07 was assembled from the CNS metagenomic data from sample V11-07, and approximately 56% of the genome of MVEV-V11-10 was assembled from the brain metagenomic data from sample V11-10. Mapping of paired end data from each of the brain samples to a database containing all viruses from vertebrate hosts available at NCBI did not identify the presence of any other virus other than the expected flaviviruses.
Discussion
An outbreak of arbovirus disease in Victoria in 2011 saw 119 confirmed arbovirus infections in horses on 103 properties and caused the death of at least 11 horses (Department of Primary Industries, Victoria, Australia; 2011, Victoria says “goodbye” to arboviruses. Available at: http://www.dpi.vic.gov.au/agriculture/pests-diseases-and-weeds/animal-diseases/vetsource/vetwatch/vetwatch-june-2011). In the current study, CNS tissue from the 11 horses from Victoria that died displaying neurological symptoms were tested for the presence of flaviviruses, and 5 tested positive for MVEV and 6 for WNVKUN. This is a significant finding given the limited reports of flavivirus isolations from horses exhibiting neurological signs in Australia3,14 over the past few decades.
Phylogenetic analysis based on a region of the E gene has shown that all of the Victorian 2011 MVEV-positive samples are highly homogeneous and cluster together with other strains of MVEV. Isolates of MVEV have been separated into 4 lineages (I–IV) based on the analysis of partial E gene sequence data. 22 Isolates of MVEV commonly found across mainland Australia are of lineage I, whereas lineages III and IV have been geographically linked to Papua New Guinea. Lineage II has not been detected outside the northeast Kimberley region of Western Australia. 22 Based on phylogenetic analysis of partial E gene sequence data, 2011 MVEV samples cluster within MVEV lineage I. In accordance with previous findings, 22 there does not appear to be any clustering of MVEV isolates based on geographic location within lineage I.
There are 2 theories relating to the emergence of MVEV in southeastern Australia: that virus reemerges from cryptic foci in southeastern Australia 5 or that it is brought down from northern foci by migrating waterbirds. 27 Based on phylogenetic analysis of the available sequence data, it is difficult to speculate which of these theories is more likely. The MVEV 2011 samples cluster with MVEV isolated in the more recent past (in the past 15 years), and many of the older lineage I strains including MVE-1-51 also cluster together. Therefore, the data could be viewed as supporting the hypothesis that viremic water birds move the virus from an endemic foci in northern Australia to other regions, thereby maintaining a single population throughout the continent. 17 In contrast, the 100% sequence identity of partial E gene sequence data from Victorian 2011 MVEV samples with 2 MVEV samples isolated from mosquitoes in NSW in 2008 indicates that the virus may have reemerged from southern foci in 2011.
WNV has been divided into 4 lineages based on whole genome comparison, which mostly correspond with geographic distribution. 28 Lineage 1 is composed of 3 clades (1a–c). Clade 1a consists of WNV isolates from Africa, Europe, the Middle East, Russia, and the Americas; clade 1b consists of WNVKUN isolates from Australia; and clade 1c consists of WNV isolates from India. 28 The phylogenetic analysis of genomic sequence data in the present study indicates that the 2011 WNVKUN samples are highly homogenous and cluster within clade 1b of WNV lineage 1. Analysis of the partial E gene of WNVKUN isolates showed the 2011 WNVKUN clustering with WNVKUN isolated in the more recent past (1991 onward). These findings, along with the similarities among the life cycle, host, vectors, and enzootic distribution of WNVKUN and MVEV, indicate that it is likely that WNVKUN has a similar dispersal and cycling patterns to that proposed for MVEV.17,27 However, as with MVEV, it is unclear whether WNVKUN in the 2011 outbreak in Victoria emerged from northern or southern foci based on phylogenetic analysis of available sequence data, and either proposal is possible.
The use of full genome sequencing for pathogen diagnosis during an outbreak situation not only identifies the virus but also allows more comprehensive characterization of the virus than can be achieved using a single gene or genome segment. The high degree of genetic homogeneity observed between isolates of MVEV and isolates of WNVKUN based on sequence analysis of individual genes indicates that, analysis during an outbreak situation, or even assessment of season to season population variation, will benefit from the increased resolving power of complete genome sequencing. However, in contrast to other flaviviruses including WNV and Japanese encephalitis virus, there is limited genomic information available for MVEV and WNVKUN. There is currently only 1 complete genome of MVEV (MVE-1-51) and 4 complete genomes of WNVKUN (MRM61C, MRM16, K6453, and WNVNSW2011) available in the NCBI database for comparison.
A genomic approach to diagnostics in an outbreak situation can also enable monitoring of the emergence of mutations that may confer virulence. Such mutations are unlikely to be identified by sequence analysis of a single gene. A study published in 2012, 12 using a WNVKUN isolate discovered during an outbreak of disease in NSW in 2011 (WNVNSW2011) has shown that, in weanling and young adult mice, this isolate is more neuroinvasive than the prototype MRM61C but not as virulent as WNV99. In addition, 2 known markers of WNV virulence were identified in the polyprotein of WNVNSW2011 that were not present in MRM61C: 12 the glycosylation site of the E-protein at residue 154 (polyprotein 446) 19 and a phenylalanine at residue 653 of NS5 (polyprotein 3181). 24 Comparison of the polyproteins of the Victorian 2011 WNVKUN isolates to other WNVKUN and WNV isolates reveals that the Victorian 2011 WNVKUN isolates are nearly identical to WNVNSW2011 (3 amino acid differences) and also include the 2 WNV virulence markers. However, it is noteworthy that the WNVKUN isolate K6543 (Australia 1991) also contains these substitutions, although the virulence of K6543, when intraperitoneally injected into 3- to 4-week-old female NIH (National Institutes of Health) Swiss outbred mice, was not significantly different to MRM16. 4 This suggests that, along with the previously identified virulence markers, 12 there are likely to be additional factors contributing to the enhanced virulence of WNVNSW2011 that differ from K6543. Further functional research is needed to determine the underlying mechanism for the enhanced virulence observed in the 2011 WNVKUN strains.
Of the 45 amino acid differences between MVEV-V11-10 and MVE-1-51, 1 was within the hinge region of the E-protein, which has been associated with virulence.21,34 While this variation may be associated with an increase in virulence of MVEV-V11-10, there is no evidence to suggest any variation in virulence between MVEV-V11-10 and MVE-1-51. Indeed, variation in this specific amino acid position has not been associated with virulence within this region. 21
Sequencing of the virus directly from host tissue using a metagenomic approach enables the unbiased amplification of the viral components regardless of predetermined ideas regarding the causal agent and culturability. For example, metagenomics was used to diagnose and differentiate swine origin H1N1 Influenza A virus in the 2009 outbreak from seasonal H3N2 or H1N1 infection 16 and to identify a novel virus (Schmallenberg virus) from the genus Orthobunyavirus in 2011 from cattle in Germany. 20 In the current study metagenomic analysis of the CNS tissue extracted from encephalitic horses successfully sequenced the entire genome of WNVKUNV11-07 and 56% of the genome of MVEV-V11-10. The viral genome sequences assembled from the CNS tissue samples V11-10 and V11-07 were identical matches to the genome sequence assembled from the respective propagated viruses from these samples. As viruses were able to be propagated from only 4 of the 11 samples tested in the current study (1 MVEV isolate and 3 WNVKUN isolates), a metagenomic analysis may provide the only means to obtain large amounts of sequence data from samples where the viral pathogen cannot be cultivated.
In addition, metagenomic analysis was able to eliminate the presence of any other viruses of similar or higher titer that may have been contributing to disease in the samples that were tested. In the event an animal is infected with an unknown or mutated virus that is unable to be detected by specific RT-PCR, or it is infected with multiple viruses, metagenomic analysis is an unbiased means of identifying all of the viral components from a single sample of tissue. Given that the cost of high throughput sequencing is steadily reducing, metagenomics is becoming a more accessible tool for comprehensive routine diagnostics.
Footnotes
Acknowledgements
The authors wish to acknowledge Xinlong Wang for technical support; the Veterinary Pathologists and the District Veterinary Officers, Department of Primary Industries (DPI), Victoria who carried out the necropsies and submitted samples for testing; and the Molecular Genetics team at the Victorian AgriBiosciences Centre, DPI, Victoria for sequencing support.
a.
Applied Biosystems MagMAX 96 Viral RNA Isolation Kit using a MagMAX Express Magnetic Particle Processor, Life Technologies Corp., Carlsbad, CA.
b.
Applied Biosystems AgPath-ID One-Step RT-PCR Kit using an Applied Biosystems 7500 Fast Real-Time PCR System, Life Technologies Corp., Carlsbad, CA.
c.
Invitrogen SuperScript III One-Step RT-PCR System, Life Technologies, Carlsbad, CA using a Bio-Rad C1000 thermal cycler, Bio-Rad Laboratories, Hercules, CA.
d.
Invitrogen Platinum Taq DNA Polymerase, Life Technologies Corp., Carlsbad, CA.
e.
Invitrogen SYBR Safe DNA Gel Stain, Life Technologies Corp., Carlsbad, CA.
f.
QIAquick PCR Purification Kit, Qiagen Inc., Valencia, CA.
g.
Australian Genome Research Facility, Parkville, Victoria, Australia.
h.
Invitrogen TURBO DNase, Life Technologies Corp., Carlsbad, CA.
i.
Promega Corp., Madison, WI.
j.
QIAamp Viral RNA Mini Kit, Qiagen Inc., Valencia, CA.
k.
TruSeq RNA Sample Preparation Guide, Illumina Inc., San Diego, CA.
l.
Finnzymes Phusion High-Fidelity DNA Polymerase, Thermo Fisher Scientific Oy, Vantaa, Finland.
m.
Library Quantification Kit–Illumina/Universal, Kapa Biosystems Inc., Woburn, MA.
n.
DNA 1000 kit run on an Agilent 2100 BioAnalyzer, Agilent Technologies, Santa Clara, CA.
o.
TruSeq PE Cluster Kit v3-cBot-HS kit run on a cBot cluster generation system, Illumina Inc., San Diego, CA.
p.
HiSeq 2000, Illumina Inc., San Diego, CA.
q.
Victorian Bioinformatics Consortium, Clayton, Victoria, Australia.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The funding for this work was provided by the Victorian Department of Primary Industries.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.