
Abstract
Canine respiratory coronavirus (CRCoV) is one of the main causative agents of canine infectious respiratory disease (CIRD), an illness whose epidemiology is poorly understood. We assessed the prevalence, risk factors, and genetic characterization of CRCoV in privately owned dogs in the Southeastern United States. We PCR-screened 189 nasal swabs from dogs with and without CIRD clinical signs for 9 CIRD-related pathogens, including CRCoV; 14% of dogs, all diagnosed with CIRD, were positive for CRCoV, with a significantly higher rate of cases in younger dogs and during warmer weather. Notably, the presence of CRCoV, alone or in coinfection with other CIRD pathogens, was statistically associated with a worse prognosis. We estimated a CRCoV seroprevalence of 23.7% retrospectively from 540 serum samples, with no statistical association to dog age, sex, or season, but with a significantly higher presence in urban counties. Additionally, the genomes of 6 CRCoVs were obtained from positive samples using an in-house developed targeted amplicon-based approach specific to CRCoV. Subsequent phylogeny clustered their genomes in 2 distinct genomic groups, with most isolates sharing a higher similarity with CRCoVs from Sweden and only 1 more closely related to CRCoVs from Asia. We provide new insights into CIRD and CRCoV epidemiology in the Southeastern United States and further support the association of CRCoV with more severe cases of CIRD. Additionally, we developed and successfully tested a new amplicon-based approach for whole-genome sequencing of CRCoV that can be used to further investigate the genetic diversity within CRCoVs.
Keywords
Canine infectious respiratory disease (CIRD) complex, colloquially referred to as kennel cough, is a significant cause of morbidity and respiratory illness in dogs, particularly prolific in the kenneled dog population. 41 CIRD is a complex disease with multifactorial etiology, where host, pathogen, and environmental interactions influence the outcome of the infection. Characterized by common clinical signs, such as coughing, nasal discharge, fever, and lethargy, CIRD can persist for several weeks, and severe bronchopneumonia can often develop. 32 The complex multifactorial etiology of this disease involves the traditional pathogens: canine parainfluenza virus (CPIV; Paramyxoviridae), 3 canine adenovirus 2 (CAV-2; Adenoviridae, Canine mastadenovirus A; https://ictv.global/taxonomy), 13 canine distemper virus (CDV; Paramyxoviridae, Morbillivirus canis), 17 canine herpesvirus (CHV; Orthoherpesviridae, Varicellovirus canid-alpha1; https://ictv.global/taxonomy), 24 and the bacteria Bordetella bronchiseptica. 5 New or emerging pathogens have also been identified as causative agents of CIRD, such as Mycoplasma cynos,32,34 canine pneumovirus (CnPnV; Paramyxoviridae), 39 canine influenza A virus H3N8 (CIV; Orthomyxoviridae, Alphainfluenzavirus influenzae), 9 and canine respiratory coronavirus (CRCoV; Coronaviridae, Betacoronavirus 1). 18
Intranasal and parenteral vaccines for a few pathogens involved in CIRD, including CPIV, CAV-2, and B. bronchiseptica, have been available for several years for routine vaccination. 10 However, outbreaks of respiratory disease occur despite their use.16,34 In Europe, a substantial proportion of dogs vaccinated against the classic CIRD agents remained clinically susceptible to respiratory disease; CRCoV was found to be highly prevalent and associated with increased severity of clinical disease. 34 A vaccine for CRCoV is not available, to date.
CRCoV was first identified in 2003 in the United Kingdom in a rescue center with a high incidence of CIRD. 18 Additional surveys have identified antibodies (Abs) in the canine population in the UK, Ireland, Italy, Japan, United States, and New Zealand.2,11,15,33,35 Several studies suggested that CRCoV contributes to the development of respiratory disease in infected dogs.14,15,23 However, not all dogs with serologic evidence of recent CRCoV infection had a clinical history of respiratory illness. Additionally, some affected dogs were coinfected with other respiratory pathogens, and establishing an association between CRCoV and respiratory disease is challenging. 15
CRCoV is a betacoronavirus (Nidovirales, Coronaviridae, Orthocoronavirinae), an enveloped virus containi-ng a non-segmented, positive-sense, single-stranded RNA genome. 42 CRCoVs are genetically distinct from canine enteric coronavirus (CECoV; Coronaviridae, Alphacoronavirus 1), the etiologic agent of enteric disease in dogs 7 ; vaccines against CECoV do not elicit protection against CRCoV infection. 15 The coronaviruses (Coronaviridae, Betacoronavirus 1) most closely related to CRCoV are human coronavirus (HCoV-OC43) and bovine coronavirus (BCoV). With the latter, CRCoV shares the highest homology (97.3% nucleotide [nt] identity). More importantly, antigenic cross-reactivity has been described between CRCoV antigen and BCoV antibodies, and vice versa. 18 The high genetic similarities between CRCoV, BCoV, HCoV-OC43, and other human enteric CoVs imply that they may have a common ancestor.26,27 A recombination event that led to strain CRCoV-K37 (Korea) 30 has been proposed between the strain CRCoV-BJ232 (China) 31 and BCoV; the spike (S) gene of CRCoV-K37 clustered with CRCoV-BJ232; however, ORF1ab, membrane (M), and nucleocapsid (N) genes were more closely related to BCoV than CRCoV-BJ232. 31 CRCoV has been suggested to be of bovine origin and to have been transmitted to dogs from cattle. 29 Although CRCoV strains frequently circulate in dogs worldwide,14,16,18,32,34 little is known about their genomic evolution, and only a few CRCoV complete genomes are available.1,30,31
We aimed to determine the prevalence and risk factors associated with CRCoV infections in dogs in the Southeastern United States and the genetic background of the CRCoV circulating in this area. We screened nasal swabs from privately owned dogs with and without CIRD for CRCoV. We also evaluated retrospectively the molecular and serologic prevalence of CRCoV, gaining information regarding CRCoV occurrence according to age, sex, geographic ubications, seasonality, and clinical signs. In addition, we obtained complete genome sequences and assessed the evolutionary genetics of CRCoV strains identified in different years in the United States and their phylogenetic relationship with CRCoV strains from Europe and Asia.
Materials and methods
Study design and origin of samples
Samples that we used for CRCoV molecular prevalence were part of regular clinical submissions between April 2018 and July 2021 to the Athens Veterinary Diagnostic Laboratory (AVDL; Athens, GA, USA). For the subclinical group (n = 154), nasal swabs were collected from privately owned dogs with clinical respiratory signs of CIRD and submitted for a canine respiratory PCR panel by licensed veterinarians practicing in the Southeastern United States. The AVDL canine respiratory PCR panel included CRCoV, CAV, CIV, CDV, CPIV, CHV, Mycoplasma canis, M. cynos, B. bronchiseptica, and Streptococcus equi subsp. zooepidemicus. Each clinical sample was accompanied by a paper submission form, which reported signalment, clinical presentation, and vaccination history, if applicable.
To investigate the prevalence of CRCoV in the absence of clinical signs of respiratory disease, nasal and pharyngeal swabs were collected postmortem from subclinical dogs (control group; n = 35) regularly submitted (March 2021–July 2021) to the AVDL for autopsy. In the control group, there was no history of CIRD, according to the submission form. Following postmortem and histologic examinations performed on the control group by a board-certified pathologist (U. Blas-Machado), the animals were confirmed to not be affected by respiratory disease.
For the CRCoV serologic investigation, we included 540 serum samples submitted to the AVDL between May 2016 and June 2021. Samples were submitted by licensed veterinarians across Georgia, South Carolina, North Carolina, Florida, and Tennessee (USA) for different reasons, including core vaccine titer screening. Similarly, when present, signalment, clinical, and vaccination history data were retrieved from the paper submission form accompanying each sample.
CRCoV detection by RT-qPCR
Total RNA was extracted (QIAamp cador pathogen kit, QIAcube automated nucleic acid extraction system; Qiagen), according to the manufacturer’s instructions, and stored at −80°C until used. The RNAs extracted were tested for the presence of CRCoV RNA by reverse-transcription quantitative real-time PCR (RT-qPCR) targeting a partial sequence of the RNA-dependent RNA polymerase (RdRp) gene (nt 14,610–nt 14,691 in the reference CRCoV strain K37 [GenBank JX860640]) as described previously, 41 with modifications. Briefly, each 25-μL PCR reaction mixture contained 12.5 μL of 2× buffer (AgPath-ID one-step RT-PCR; Qiagen), 3 μL of each primer (0.4 μM) and 3 μL of the TaqMan probe (0.4 μM), 1 μL of enzyme mix, and 8 μL of template RNA. The following temperature cycling protocol was used: reverse transcription at 45°C for 10 min and denaturation at 95°C for 10 min, followed by 40 cycles of 15 s of denaturation at 95°C, 45 s of primer annealing, and elongation at 55°C. All RT-qPCRs were performed with a positive amplification control (plasmid containing the sequence of interest; Genewiz), a negative amplification control (nuclease-free water), and exogenous internal controls (QuantiFast pathogen internal control kit; Qiagen).
A targeted amplicon approach for CRCoV whole-genome sequencing
To maximize the next-generation sequencing (NGS) success of CRCoV, only CRCoV RT-qPCR–positive samples with a cycle threshold (Ct) value ≤ 28 were selected for sequencing. A multiplex PCR primer scheme for target amplicon sequencing of CRCoV was designed utilizing the web-based primer design tool Primal Scheme (http://primal.zibraproject.org/), and the complete genome sequence of CRCoV-K37 (GenBank JX860640) as reference strain, specifying an amplicon length of 400 bp with an overlap of 0 bp (Suppl. Tables 1, 2). The lyophilized primers (Integrated DNA Technologies) were resuspended in nuclease-free water to achieve a stock concentration of 100 μM, according to the manufacturer’s recommendations. We generated 2 primer pools by combining 5 μL of each primer, followed by a dilution of each pool in nuclease-free water to produce 10 μM stocks, with each primer used at a final concentration of 0.015 μM for the multiplex RT-qPCR. cDNA synthesis was performed using 2 μL of Luna Script RT mix (New England Biolabs) and 8 μL of RNA. This reaction was incubated at 25°C for 2 min, 55°C for 20 min, 95°C for 1 min, then held at 4°C. For the amplicon-based multiplex RT-qPCR, 2 reactions per sample were set up by combining 2.52 μL of nuclease-free water, 12.5 μL of master mix (Q5 Hot Start High-Fidelity 2× master mix; New England Biolabs), 3.98 μL of each primer pool, and 6 μL of cDNA. The thermocycling conditions were as follows: 1 cycle of 98°C for 30 s, followed by 35 cycles of 95°C for 15 s and 65°C for 5 min, and held at 4°C. Amplicons generated from the 2 multiplex PCRs were combined to obtain a total volume of 25 μL and cleaned up (1× AMPure XP beads; Beckman Coulter). The pellet was resuspended in 26 μL of nuclease-free water, and 1 μL of the eluate was used to determine the DNA concentration (Qubit double-stranded DNA assay kit; Thermo Fisher). Sequencing libraries were made (Nextera DNA flex library preparation kit; Illumina) following the protocol described previously. 37 Paired-end sequencing was performed (MiSeq reagent kit v.3, 300 cycles, MiSeq instrument; Illumina).
Bioinformatic analysis
Illumina paired-end reads were first checked qualitatively with FastQC v.0.11.8 45 (Illumina) and then submitted to Trimmomatic v.0.36 6 (Illumina) to remove adapters and select reads with Phred scores < 30. De novo assembly was performed with SPAdes v.3.15, 4 followed by scaffolding with RagTag v.2.1 1 and CRCoV-K37 (GenBank JX860640) 29 as the reference genome. The assembled genomes were submitted to NextPolish 1.4 21 for error correction and assembly base-call polishing. The complete genome sequences were annotated using a pipeline for prokaryotic and viral genomes, PROKKA. 40 CRCoV USA 1–5 genome sequences were deposited in GenBank (ON133845–ON133848, ON133844). Partial nucleotide sequences of M and ORF1ab genes of CRCoV USA 6 were deposited in GenBank (ON072523 and ON072524, respectively). To determine the evolutionary relationships between the CRCoV that we discovered and those identified previously, all 4 homologous publicly available CRCoV complete genome sequences, along with 7 and 8 complete genome sequences of the most related alphacoronavirus and betacoronavirus members, respectively, were recruited from GenBank and the European Nucleotide Archive at EBI (Suppl. Table 3). Nucleotide sequence alignment was performed using the Multiple Alignment Fast Fourier Transform (MAFFT) software 25 within Geneious Prime v.2022.1.1 (Biomatters). A maximum-likelihood (ML) phylogenetic tree was constructed using MEGA X, 28 implementing the general time reversible model as a substitution model, 36 and 1,000 bootstrap replicates.
Additionally, 15 S gene sequences (coding for the spike protein) of CRCoVs for which the complete genome sequences were not available (Suppl. Table 4) were recruited from GenBank and the European Nucleotide Archive at EBI. Within these additional 15 sequences, 7 were complete S gene sequences; 8 were partial (nt 24,837–27,244 of CRCoV-K37 [GenBank JX860640]). All of these 15 S gene sequences, along with the S gene sequences extracted from the whole genome alignment, were aligned as described above, and a partial S gene ML phylogenetic tree was constructed using MEGA X, 28 implementing the general time reversible model as substitution model, 43 and 1,000 bootstrap replicates. Thirty-two complete S gene sequences (24 extracted from the whole genome alignment and 8 of the additional 15 mentioned above) were subsequently translated into amino acids in Geneious Prime v.2022.1.1. The amino acid sequences were aligned with MAFFT, and a heatmap containing the percentage similarity of the spike protein sequences was generated (Prism v.9.3.1; GraphPad). The spike protein amino acid alignment was also used to create a phylogenetic tree using the ML method and the standard Whelan and Goldman (WAG) substitution model, 43 implemented in MEGA X. 28
Serosurveillance of CRCoV
We determined CRCoV antibodies in 540 canine sera included using a commercial competitive ELISA coated with a purified BCoV (BIO K 392-monoscreen antibody ELISA bovine coronavirus/competition; Bio-X), performed according to the manufacturer’s instructions using provided positive and negative controls. Briefly, serum samples were diluted 1:20 in the dilution buffer, and 100 μL of each were dispensed in duplicate into wells, proceeding in the same manner for the reference sera (positive and negative sera). Then, the conjugate was diluted 20-fold in the dilution buffer, and 100 μL dispensed into each well and incubated at 21°C for 1 h. Following 3 washes, 100 μL of the chromogen solution was added to each well on the plate, and incubation was performed for 10 min at 21°C. The reaction was stopped with 50 μL of stop solution per well. The results were calculated based on the optical density at 450 nm (OD450) and described as a percentage of inhibition (POI). 35 According to the recommended instructions, samples with POI ≥ 20% were considered positive for CRCoV antibodies.
Statistical analysis
History, signalment, and clinical presentation data were retrieved for each animal from the sample submission form accompanying each sample received at AVDL. The following variables were extracted or estimated: sex, age, age group, severity of the disease, season (hot or cold) at the time of collection, and geographic area where the dog lived (urban or rural). Age was presented in months, and dogs were classified into different age groups (puppy, 0–5 mo; juvenile, 6–12 mo; young adult, 13–24 mo; mature adult, 25–72 mo; senior, 73–132 mo; geriatric, >133 mo) based on a previous publication. 20 The severity of the disease was classified as follows: subclinical dogs sampled at autopsy (absence of clinical, macroscopic, and histologic signs of respiratory infections), dogs with mild clinical disease (upper respiratory tract clinical signs), and dogs with moderate-to-severe clinical disease (systemic clinical signs and lower respiratory tract clinical signs).
Seasonality was divided into hot and cold based on data presented by the National Weather Service (https://www.weather.gov/ffc/) for Georgia during the years of sample collection. First, the annual average temperature for Georgia was calculated based on the average monthly temperatures of 10 cities across the state (Athens, Atlanta, Columbus, Macon, Cartersville, DeKalb, Fulton, Gainesville, Peachtree City, Rome). Months with average temperatures above the year average (May–October) were classified as hot season; months with temperatures below the year average (November–April) were classified as cold season. Geographic area was divided into urban and rural based on the census criteria to classify counties in each state.
For descriptive analysis of the data, categorical variables (sex, season, geographic area) were summarized in contingency tables; the continuous variable age was summarized using the median and ranges. For the inferential analysis, the association of the binary outcome of whether a dog was found to be CIRD- or CRCoV-positive or to carry antibodies against CRCoV with the individual dependent variables mentioned above was analyzed using logistic regression (Prism v.9.3.1). The association between CIRD pathogen and clinical respiratory sign severity was assessed using the chi-square test (Prism v.9.3.1).
Results
Prevalence of CRCoV infection in dogs suffering from CIRD
To assess the molecular prevalence, epidemiologic data were obtained from 154 dogs with clinical respiratory signs and 35 dogs unaffected by respiratory disease. Comparable proportions (~50%) of samples were collected from male and female dogs. Most animals originated from Georgia, with ~10% from North Carolina, South Carolina, Florida, and Texas. Estimated age was recorded for each dog and ranged from 1-wk- to 15-y-old, with 41 puppies, 32 juvenile dogs, 35 young adult dogs, 33 mature adult dogs, 27 senior dogs, and 13 geriatric dogs. Sampled dogs inhabited urban counties (163 of 189, 86%); only 26 of 189 (14%) inhabited rural regions. In addition, 119 of 189 (63%) samples were collected during the hot season; 70 of 189 (37%) were collected during the cold season. By logistic regression analysis, age (p = 0.028) and season of collection (p = 0.031) were significantly associated with increased odds of identifying dogs suffering from CIRD (Table 1).
Association of CIRD cases (positive/negative) and CRCoV cases (positive/negative) with dog’s sex, age, season (hot/cold) of sample submission, and county (urban/rural) of residence.
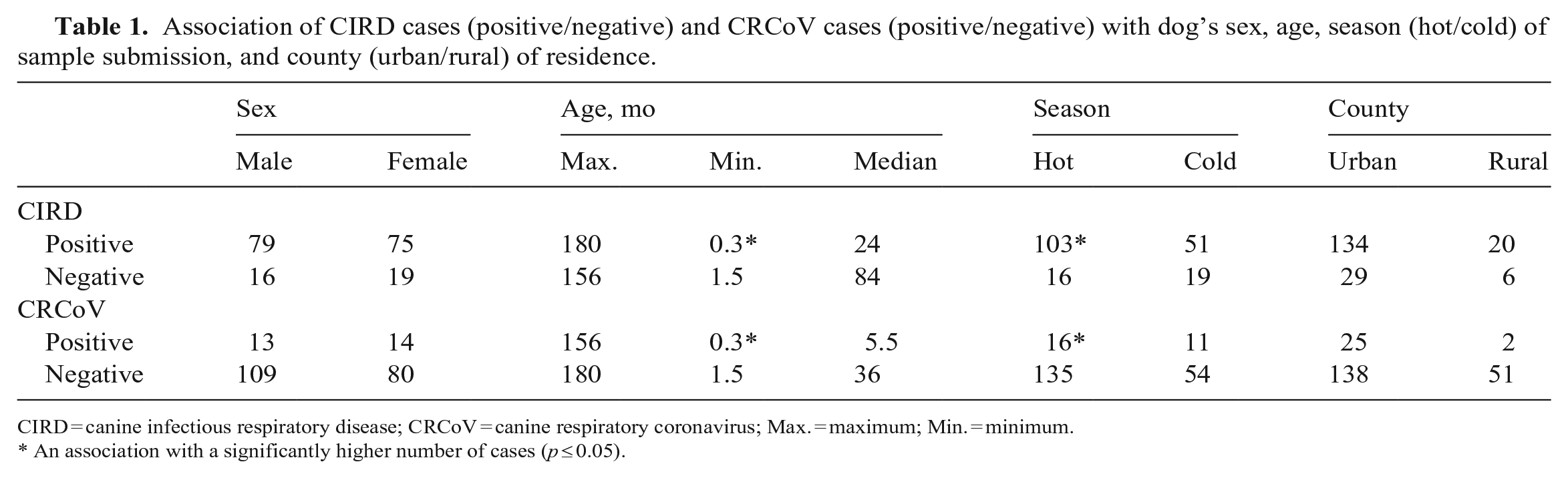
CIRD = canine infectious respiratory disease; CRCoV = canine respiratory coronavirus; Max. = maximum; Min. = minimum.
An association with a significantly higher number of cases (p ≤ 0.05).
In our AVDL canine respiratory PCR panel, 74 of 154 (48%) reported CIRD cases were caused by an unknown pathogen. In contrast, most CIRD cases with known etiology were caused by Mycoplasma spp. (17.5%), followed by polymicrobial infections (16.2%), CRCoV (9.7%), and B. bronchiseptica (5.2%; Table 2). None of the healthy dog carcasses tested positive for CRCoV or other CIRD-related pathogens. Of the 154 dogs sampled with clinical signs, only 27 were positive for CRCoV (Suppl. Table 5). CRCoV was detected by RT-qPCR in 27 of 189 dogs (14.4%; 95% CI: 10–20.0%; Ct: 21–36). Only the dogs’ age (p = 0.02) and season of collection (p = 0.005) were significantly associated with increased odds of identifying dogs infected with CRCoV (Table 1). CIRD and CRCoV infections seemed to be more common among younger dogs and during the warmer months.
CIRD causative agents detected in dogs with respiratory clinical signs.

B. bronchiseptica = Bordetella bronchiseptica; CAV = canine adenovirus; CDV = canine distemper virus; CHV = canine herpesvirus; CIRD = canine infectious respiratory disease; CPIV = canine parainfluenza virus; CRCoV = canine respiratory coronavirus; S. equi subsp. zooepidemicus = Streptococcus equi subspecies zooepidemicus.
Within the group of dogs with CIRD, 62 of 154 (40%) were mildly affected clinically, with only upper respiratory signs, such as sneezing and coughing. In comparison, 94 of 154 (60%) had systemic clinical signs, including fever, lethargy, and lower respiratory tract disease. Clinical signs were moderate-to-severe in 24 of the 27 dogs diagnosed with CRCoV infection. The detection of CRCoV from the clinical samples submitted for the canine respiratory PCR panel was statistically associated with moderate-to-severe clinical signs compared with the infection rates in the group with mild clinical signs (p = 0.003 and p = 0.001, respectively; Fig. 1A). Notably, a further statistically significant result was obtained when CRCoV was the only CIRD pathogen detected (moderate-to-severe disease group vs. subclinical group, p = 0.032; moderate-to-severe disease group vs. mild disease group, p = 0.020; Fig. 1B).
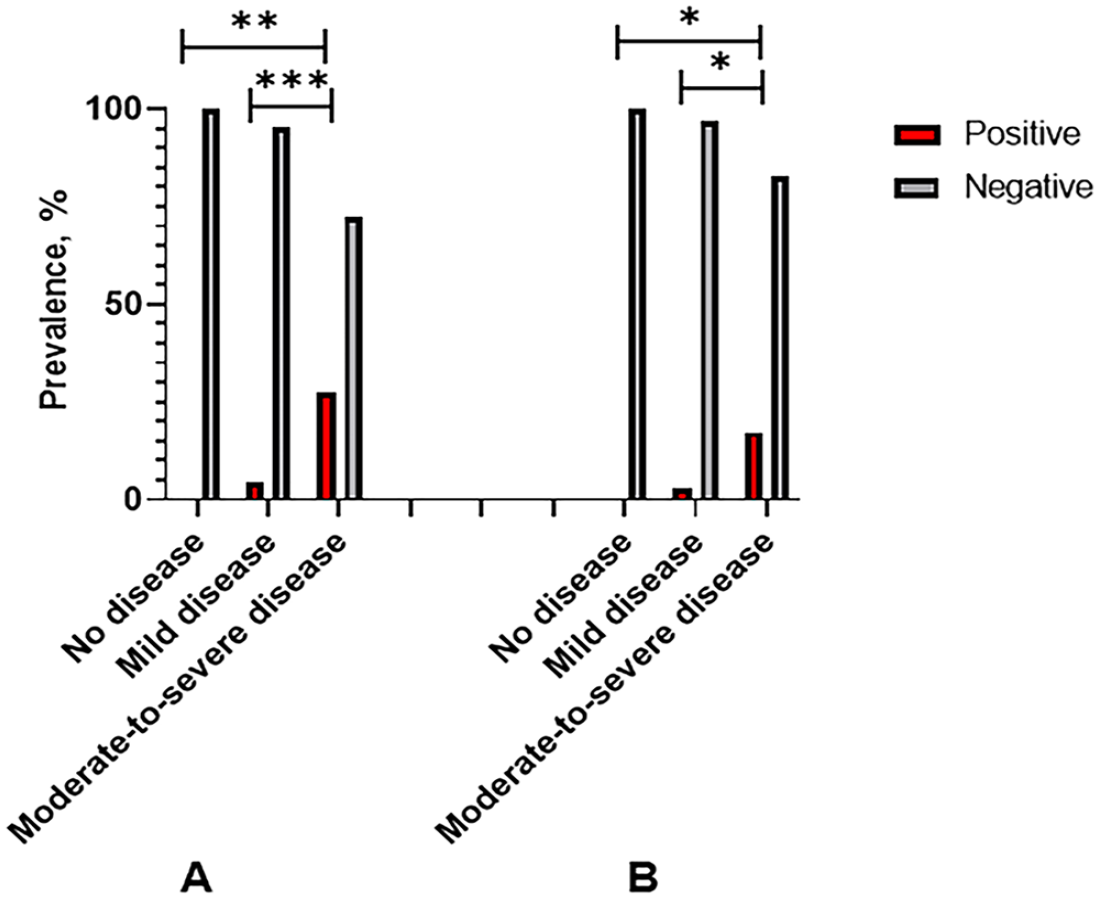
Rate of molecular detection of pathogens by clinical presentation:
Genome characterization and phylogeny of 5 CRCoV strains isolated in the United States
Whole-genome sequencing (WGS) generated 3,298,188 raw reads (median: 601,368 reads per sample), of which 3,219,885 (median: 594,761.5 reads per sample) passed the quality filter and were used for genome assembly (Table 3). De novo assembly and scaffolding generated a single contig of ~30,000 bp for 5 of 6 samples that we sequenced, which is in line with the size of the CRCoV reference genome strain K37 (GenBank JX860640). Each contig was an almost-complete CRCoV genome with a genomic organization analogous to the one noted in the reference genome, CRCoV-K37. All 5 genomes lacked the entire 5′UTR region, the first 9 nucleotides of the ORF1a and ORF1ab sequences, and the final 3′UTR region (Suppl. Fig. 1). The genome organization of these 5 isolates was typical of betacoronavirus, with 12 coding regions predicted, including the two-thirds occupied by the replicase gene in the 5′-end, comprised of 2 overlapping ORFs called 1a (nt 1–13,143) and 1b (nt 1–21,275; Suppl. Fig. 1). Other coding sequences predicted in the order in which they were found in the genome were: HE (nt 22,133–23,407), S (nt 23,422–27,513), E (nt 28,203–28,457), M (nt 28,472–29,164), NP (nt 29,174–30,520), N (nt 29,235–29,858), along with 4 genes encoding nonstructural proteins of 32 kDa (nt 21,285–22,121), 4.9 kDa (nt 27,503–27,637), 2.7 kDa (nt 27,673–27,750), and 12.8 kDa (nt 27,887–28,216). Only 2 partial gene sequences were obtained from 1 sample (CRCoV USA 6): 693-bp (M gene) and 1,587-bp (ORF1ab gene) sequences (nt 28,691–29,383 and nt 225–1,811 of the reference genome CRCoV-K37, respectively).
Assembly and genomic features of the 6 CRCoV strains that we identified.
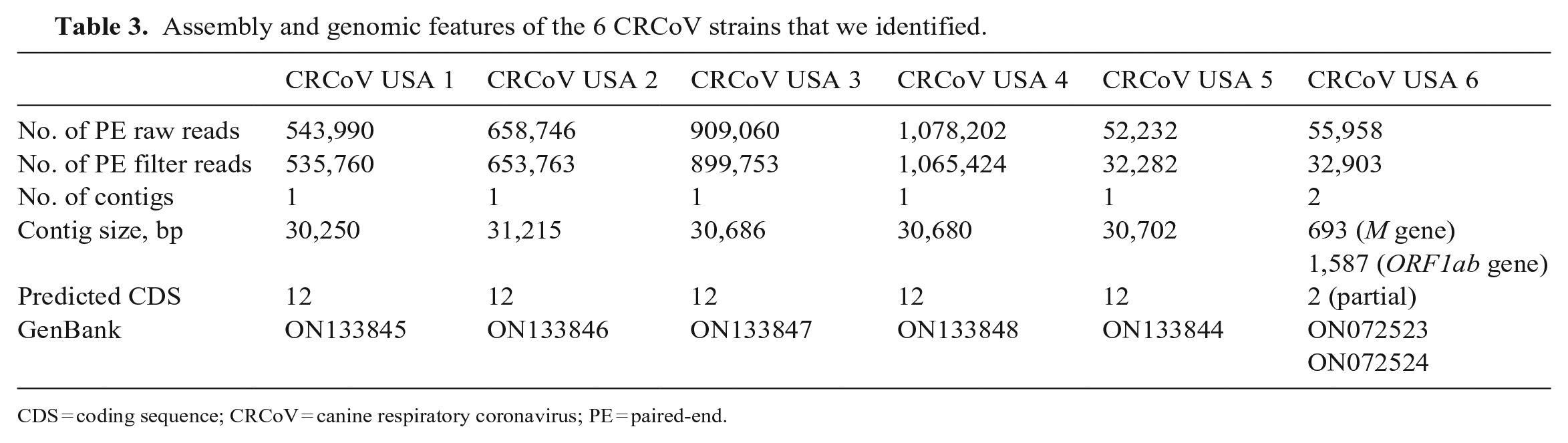
CDS = coding sequence; CRCoV = canine respiratory coronavirus; PE = paired-end.
To understand the genetic variation within CRCoV samples from our study and other CRCoV strains available, nucleotide sequence identity analyses were performed for individual genes as described previously. 44 ORF1ab, S, HE, and M genes had 97.3–100% genetic similarities with CRCoV strains identified in the Southeastern United States. Similarly, based on the genome sequences, the nt sequence identity values were 99.4% within CRCoVs USA 1–3. In contrast, CRCoVs USA 4 and 5 were slightly distant, sharing 98.8% nt identity with the other strains in our study. All CRCoV USA strains, except for CRCoV USA 4, had the highest nt sequence identity with CRCoV strains from Sweden, CRCoV S4 and CRCoV S11 (GenBank PRJEB34079), 44 with nt identity of 99.2–99.6%, 98.2–99.4%, 98.5–99.3%, 99.1–99.3%, and 99.0–99.4% for the genes ORF1ab, HE, M, and S, and genome sequences, respectively. On the other hand, CRCoV USA 4 bore the highest nt identity with CRCoV strains BJ232 (GenBank KX432213.1) and K37 (GenBank JX860640), isolated in China and Korea, respectively,29,31 with nt identity ranges of 98.5–99.8%, 97.6–99.2%, 97.5–99.7%, 99.5–99.6%, and 98.6–99.7% for the genes ORF1ab, HE, M, and S, and genome sequences, respectively. It is well known that given the redundancy of the genetic code, changes of nt sequences are accumulated at a higher rate than those in amino acid sequences and therefore protein sequences, conserving a better phylogenetic signal, and are used more commonly to infer phylogenetic relationships. The nt sequences of the genes ORF1ab, HE, M, S, and genome sequence were translated, and the comparison of amino acid (aa) sequences confirmed that most of our CRCoV strains shared the highest aa sequence identity (99.5–99.1%, 99–97.4%, 98.7–97.8%, 99.4–98.6, and 99.0–99.3%, respectively) with CRCoV strains isolated in Sweden, with the consistent exception of CRCoV USA 4, which was most closely related to CRCoV strains from Asia (99.7–98.9%, 98.6–96.9%, 99.6–97.4%, 99.3–99.0%, 99.4–97.4%, respectively; Fig. 2).
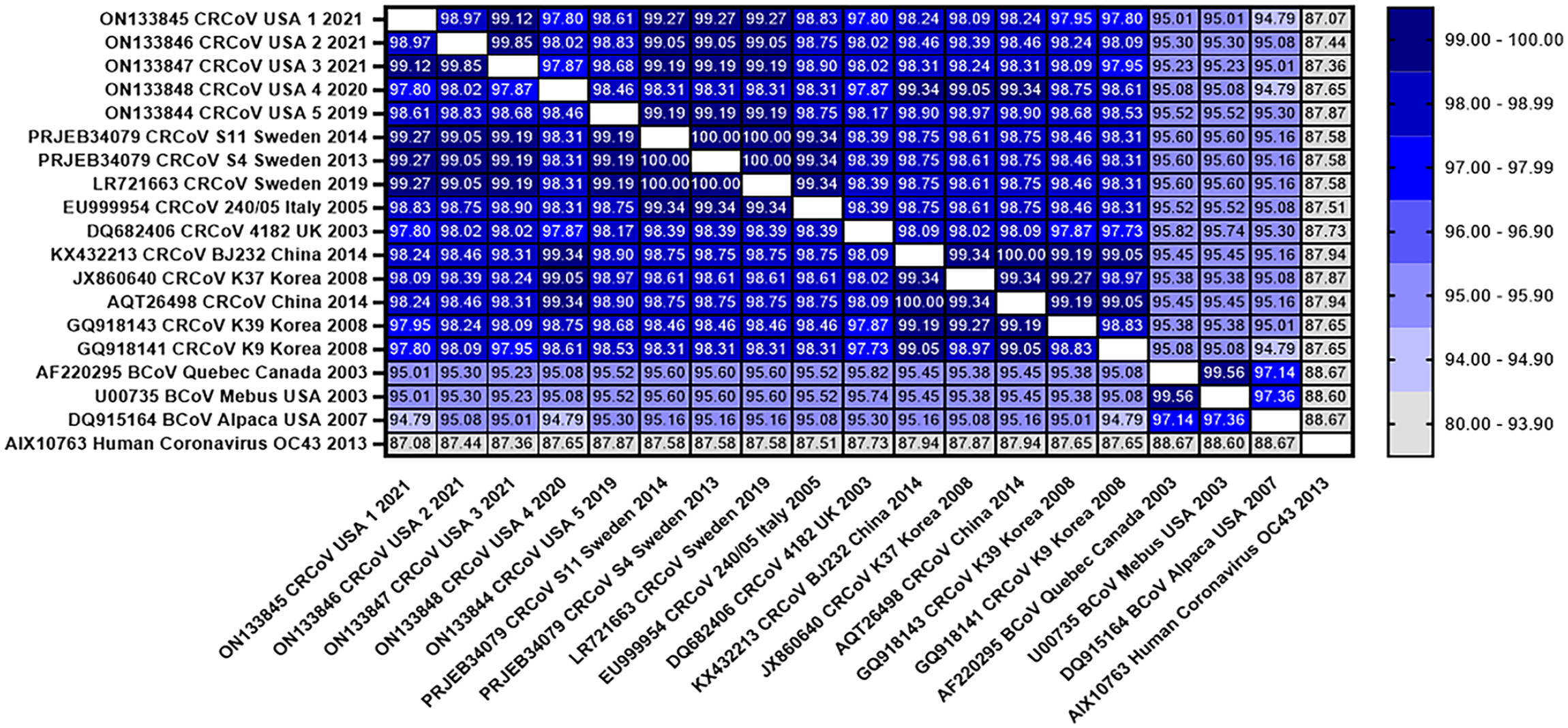
Heatmap of the percentage identity between canine respiratory coronavirus (CRCoV) and the closest related betacoronaviruses (BCoVs; Quebec, Mebus, Alpaca) and human coronavirus (HCoV) strain OC43. The heatmap was generated based on the amino acid similarity of the spike protein (GraphPad Prism v.9.3.1). The color key on the right indicates the percent protein similarity.
As expected, CRCoV strains from our study belonged to the betacoronavirus group, and they shared a common origin with BCoV strains (Fig. 3). CRCoV 1/2021/USA, CRCoV 2/2021/USA, CRCoV 3/2021/USA, and CRCoV 5/2019/USA strains (our study) clustered with CRCoV S11/2014/Sweden and CRCoV S4/2013/Sweden, 44 separated from the group formed by CRCoV 4/2020/USA (our study), CRCoV BJ232/2014/China, 29 and CRCoV K37/2008/Korea. 31 The resulting phylogenetic trees based on the S gene (Suppl. Fig. 2A) and on the S protein (Suppl. Fig. 2B) had the same cluster assignment, confirming that CRCoV strains are divided into 2 clusters: cluster I, containing the 4 CRCoV strains from our study and those from Sweden and Italy; and cluster II, including 1 from our study and those from China, Korea, and the UK.

Phylogenetic analysis is based on our 5 whole-genome sequences and 19 representative coronaviruses. The evolutionary history was inferred using the maximum-likelihood method and general time reversible model. 33 The percentage of trees in which the associated taxa clustered is shown next to the branches. A discrete gamma distribution was used to model evolutionary rate differences among sites (5 categories [+G, parameter = 2.5701]). The rate variation model allowed some sites to be evolutionarily invariable ([+I], 15.4% sites). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. GenBank accessions are indicated in each branch. The gray and colored branches indicate members of the genera Alphacoronavirus and Betacoronavirus, respectively (blue = those from Sweden and 4 canine respiratory coronaviruses [CRCoVs] from the United States [cluster I]; red = those from the UK and Asia, along with CRCoV 4/2020/USA [cluster II]; purple = betacoronaviruses [BCoVs]).
Serosurveillance of CRCoV detected significantly higher levels of CRCoV antibodies in urban counties
We assessed retrospectively the seroprevalence of CRCoV antibodies in 540 canine sera samples. There were similar numbers of males and females (252 males, 288 females); the maximal and minimal ages were 192 and 2 mo (median: 72 mo), respectively (Table 4). The origin of the 540 samples by geographic region was as follows: 500 (92.6%) were submitted from Georgia, 16 (3%) from South Carolina, 9 (1.7%) from Florida, and 15 (2.8%) from North Carolina; 415 dogs (76.9%) lived in urban counties, whereas 125 (23.1%) lived in rural counties. More than half of the samples were collected during the hot season (363 of 540; 67.2%); 177 dogs were sampled during the cold season (Table 4). We found CRCoV antibodies in 128 sera (23.7%; 95% CI: 20.1–27.3%), with no statistical differences among the different age categories or with the sex of the dogs. However, seroprevalence was significantly higher in urban counties than in rural environments (p = 0.004; Table 4).
Association between the presence of CRCoV antibodies (positive/negative) and dog’s sex, age, season (hot/cold) of sample submission, and geographical area (urban/rural).
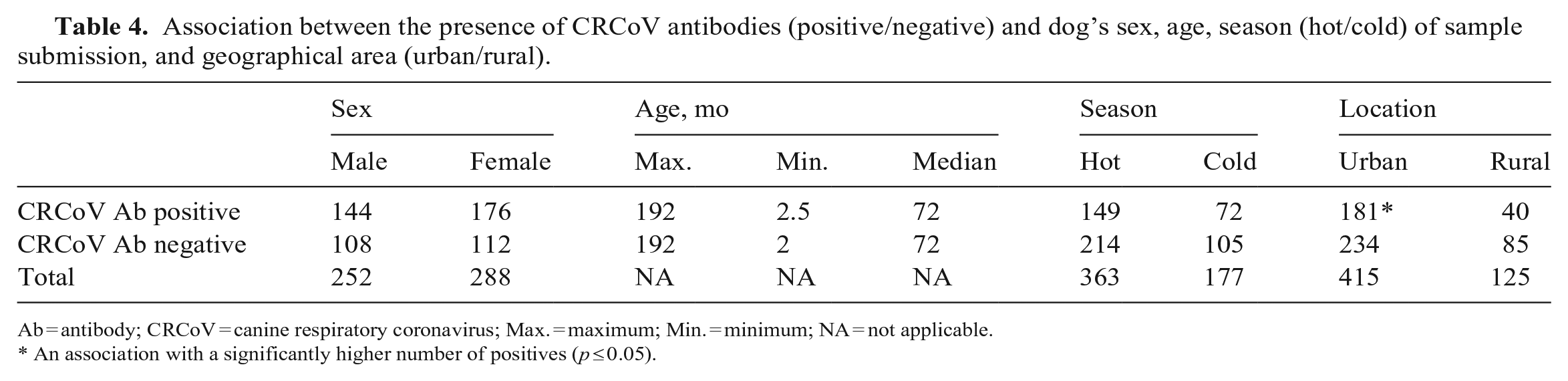
Ab = antibody; CRCoV = canine respiratory coronavirus; Max. = maximum; Min. = minimum; NA = not applicable.
An association with a significantly higher number of positives (p ≤ 0.05).
Discussion
Mycoplasma spp. (17.5%), polymicrobial infections (16.2%), CRCoV (9.7%), and B. bronchiseptica (5.2%) represented the pathogens most detected in our study; approximately half (74 of 154) of the CIRD cases were caused by an unknown pathogen. The Mycoplasma spp. detection rate that we found is consistent with a previous survey from our group, in which M. canis (23.6%) and M. cynos (24.5%) were the most-detected bacterial pathogens, followed by CPIV (29%), CIV (11.2%), and B. bronchiseptica (9%). 32 A less-apparent involvement of M. cynos and CIV was reported from Europe, 34 accompanied by a higher detection of CRCoV (7.7%) and CnPnV (23.4%). Additionally, the absence of CRCoV in the healthy control group in our study further supports the association of CRCoV with CIRD.
Vaccination against key CIRD pathogens might reduce the disease occurrence and severity in some individuals. 33 Nevertheless, a high prevalence of CIRD pathogens (201 of 306 dogs; 65.7%) was reported in a European surveillance study of dogs vaccinated against the classic CIRD agents. 34 Overall, we found that CRCoV was found exclusively in 14.3% of dogs diagnosed with CIRD, further demonstrating the relevance of this virus in the complex etiology of CIRD; this molecular prevalence rate is higher than the 4.6% rate that we found in a previous investigation of the role of coinfections of viruses and bacteria in the etiology of CIRD. 32 This difference may be a result of the methodology used; we had further optimized a RT-qPCR assay developed for CRCoV RNA detection 41 and used that test for molecular detection of CRCoV, rather than the standard consensus coronavirus RT-PCR assay used previously. 19 Importantly, we found CRCoV in dogs of different breeds and found no difference in prevalence regardless of geographic location or sex of affected dogs. However, dog age and season were consistently associated with CIRD incidence and CRCoV prevalence, evidence that CIRD infections in general and CIRD caused exclusively by CRCoV are more common among younger dogs and during the warmer months of the year. The observed seasonality of CIRD and CRCoV could be caused by fewer tests being undertaken during the cold months, and thus this association requires further investigation.
Nevertheless, the association of CRCoV disease with a younger age is consistent also with findings in Europe 34 ; a study reported that younger dogs (less likely to have antibodies to CRCoV) were more likely to suffer CIRD and be infected with CRCoV, and developed more severe clinical signs. Furthermore, although in our study CRCoV was detected in dogs with respiratory disease ranging from mild to moderate and severe with systemic involvement, CRCoV was positively associated with increased severity of the clinical disease, as observed in other studies. 34 Our findings identify CRCoV as one of the main causative agents of CIRD in pet dogs and highlight the importance of updating and developing molecular techniques and novel vaccines, especially in light of the great proportion of dogs vaccinated but still affected by respiratory disease.
Even though the recent SARS-CoV-2 pandemic significantly increased the research and WGS of coronaviruses (CoVs), only a few publicly available CRCoV complete genomes exist. Indeed, before our study, there were no genomic sequences of North American CRCoVs in public databases, with all available genomes originating from Asia and Europe. We identified complete genome sequences from 5 CRCoV isolates from the United States and used them to assess their genomic evolution and phylogenetic relationships with other CRCoV strains. Considering the success of the ARTIC protocol for the sequencing of SARS-CoV-2,12,22 we followed a similar targeted-sequencing approach 8 as a guide for the design and implementation of WGS of our CRCoV-positive samples. Overlapping primers targeting the CRCoV reference genomes were used to PCR-amplify overlapped fragments that were then sequenced and assembled to obtain the full complete CRCoV genome directly from positive samples.
The CRCoV strains that we sequenced clustered within the betacoronavirus genus and were distantly related to members of the alphacoronavirus genus, which includes CECoVs, HCoV-NL63, and HCoV-229E.30,31,44 The closest relatedness that we observed was with other CRCoV and BCoV genomes, thus corroborating findings that suggest that CRCoV originated as a host variant of BCoV or that both viruses originated from a common ancestor. 42 Four of the complete genomes from our study were closely related to the sequenced CRCoV strains from Sweden in all phylogenetic analyses performed. Additionally, despite being detected in different dogs and across different years, slight genetic variation was detected among these 4 USA CRCoV strains. This suggests that these viruses may have derived from a common introduction to the United States from Europe. One U.S. strain appeared to be slightly distant given that it shared 98.8% nucleotide identity with the other strains of our study, suggesting a closer genetic relatedness to CRCoVs from Asia. Therefore, we could separate the North American strains into 2 clusters, one including 4 of 5 strains from our study (except CRCoV 4/2020/USA) and those isolated from Sweden, 44 and the other one including CRCoV 4/2020/USA and those from China, Korea, and the UK.15,18,29,31
Finally, our serosurveillance results provide evidence that CRCoV infection was common among dogs in the Southeastern United States in 2018–2021. Overall, 23.7% of sera tested were positive for CRCoV antibodies. This was lower than the 54.5% value reported in the United States in 2006, 38 in which samples were collected mostly from urbanized areas, and lower than the 36% and 32.5% values reported in the UK and Ireland, respectively, in 2006. 38 This value was also lower than those reported by a surveillance study conducted in several European countries between 2011 and 2013, 34 which found an overall seroprevalence of 47%. It is essential to point out that the CRCoV seroprevalence was correlated with the population density given that we found higher seroprevalence in urban counties than rural counties, consistent with another study in which lower CRCoV antibody levels were detected in the regions of the southwest of England or Wales than in dogs in urban areas. 38
Supplemental Material
sj-pdf-1-vdi-10.1177_10406387231213662 – Supplemental material for Epidemiologic investigation and genetic characterization of canine respiratory coronavirus in the Southeastern United States
Supplemental material, sj-pdf-1-vdi-10.1177_10406387231213662 for Epidemiologic investigation and genetic characterization of canine respiratory coronavirus in the Southeastern United States by Eliana De Luca, Sonsiray Álvarez-Narváez, Rodrigo P. Baptista, Grazieli Maboni, Uriel Blas-Machado and Susan Sanchez in Journal of Veterinary Diagnostic Investigation
Footnotes
Acknowledgements
We thank the Molecular Biology Section of the Athens Veterinary Diagnostic Laboratory, especially Ingrid Fernandez, for her outstanding RT-PCR optimization skills, and Paula Bartlett for her exceptional guidance throughout NGS.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Our work was funded by the FDA and Vet-LIRN as part of an ongoing infrastructure grant. Eliana De Luca and Grazieli Maboni were supported by a grant from Boehringer Ingelheim administered through the College of Veterinary Medicine at the University of Georgia.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.