
Abstract
Calf diarrhea results in significant economic loss and is caused by a variety of pathogens, including enteric viruses. Many of these viruses, including bovine norovirus (BNoV), bovine torovirus (BToV), and bovine kobuvirus (BKoV), are recognized as the causative agents of diarrhea; however, they remain understudied as major pathogens. We developed a multiplex reverse-transcription quantitative real-time PCR (RT-qPCR) assay for rapid and simple detection of BNoV, BToV, and BKoV. Our method had high sensitivity and specificity, with detection limits of 1 × 102 copies/μL for BNoV, BToV, and BKoV, which is a lower detection limit than conventional RT-PCR for BNoV and BKoV and identical for BToV. We tested fecal samples from 167 diarrheic calves with our multiplex RT-qPCR method. Viral detection was superior to conventional RT-PCR methods in all samples. The diagnostic sensitivity of the multiplex RT-qPCR method (100%) is higher than that of the conventional RT-PCR methods (87%). Our assay can detect BNoV, BToV, and BKoV in calf feces rapidly and with high sensitivity and specificity.
Diarrhea in calves can increase medication costs, delay growth, and cause mortality. 6 Several viral pathogens have been associated with calf diarrhea; these pathogens can be classified as major pathogens (bovine rotavirus, coronavirus, bovine viral diarrhea virus) or minor, but increasingly important, pathogens (bovine norovirus, torovirus, kobuvirus).4,5,24
Although several studies have examined the association between major causative agents and diarrhea in bovids, comparatively little research has been conducted on the association between minor causative agents and diarrhea.1,2,22 Minor pathogens have been detected in feces of both healthy and sick animals. Field evidence associating some of these agents with calf diarrhea remains limited. Bovine norovirus (BNoV; Caliciviridae, Norovirus), bovine torovirus (BToV; Coronaviridae), and bovine kobuvirus (BKoV; Picornaviridae, Aichivirus B) may all be associated with calf diarrhea. 9
BNoVs are non-enveloped, positive-sense, single-stranded RNA viruses. 6 Noroviruses can be classified into 6 genogroups (GI–GVI) based on the main capsid protein (VP1) sequence; genogroup III (GIII) is associated with bovine infections.11,17 GIII consists of genotypes 1 (GIII.1) and 2 (GIII.2). BNoV GIII.2 causes persistent diarrhea and prolonged fecal shedding without intestinal lesions; GIII.1 leads to severe atrophic jejunitis. 16
BToVs are enveloped, positive-sense, single-stranded RNA viruses. 9 These viruses cause mild-to-moderate diarrhea in calves < 3-wk-old. 13 When BToV infects intestinal villi, it causes cell death and epithelial detachment that results in necrosis in the large intestine.7,21 BToV infection leads to malabsorptive, maldigestive, and hypersecretory diarrhea. 26
BKoVs are non-enveloped, positive-sense, single-stranded RNA viruses. 12 These viruses contribute to diarrheal diseases of cattle and the pathogenesis of calf enteritis.9,14 The infected villus epithelium of the jejunum is largely sloughed, and the lamina propria is infiltrated by inflammatory cells, causing diarrhea in cattle. 25
According to data released by the Animal and Plant Quarantine Agency in South Korea, the distributions of microorganisms detected from cases of domestic calf diarrhea from November 2013 to October 2016 were as follows: of the 17 pathogens tested (Clostridium perfringens A–C, coccidia, Giardia, rotavirus, Cryptosporidium, pathogenic Escherichia coli, norovirus, torovirus, Clostridium difficile, coronavirus, bovine viral diarrhea virus [BVDV], parvovirus, adenovirus, kobuvirus, and Salmonella spp.), norovirus accounted for 10.7%, torovirus for 7.2%, and kobuvirus for 2.3% of cases. Overall, this proportion is comparable to the percentage of cases associated with the most common viral agent, rotavirus (19.4%). These viruses can induce bovine infectious diarrhea through single or mixed infections. 15 However, the importance of viral pathogens such as BNoV, BToV, and BKoV, and their role in disease, remains unclear. Therefore, developing a method capable of simultaneous detection with high sensitivity and specificity would aid in better understanding the epidemiology, ecology, and pathogenesis of these viruses.
To date, the methods for detection of BNoV, BToV, and BKoV are reverse-transcription PCR (RT-PCR) and nested PCR assays.8,19,20,27 Considering the run time of a conventional PCR machine (Applied Biosystems ProFlex 3 × 32-well PCR system; Thermo Fisher), these techniques take 2.5 h for BNoV, 6 h for BToV, and 6.5 h for BKoV. Including a capillary electrophoresis system (QIAxcel; Qiagen), the overall method takes up to 7 h, making testing laborious and time-consuming. Therefore, developing a multiplex reverse-transcription quantitative real-time PCR (RT-qPCR) assay that could detect BNoV, BToV, and BKoV quickly, simply, and accurately would be very useful. We, therefore, aimed to develop a multiplex RT-qPCR for the detection of BNoV, BToV, and BKoV.
Materials and methods
Designing primers and probes for our RT-qPCR assay
We designed new sets of primers and probes for the detection of BNoV, BToV, and BKoV using Primer Express 3.0 software (Thermo Fisher). The primers were generated using multiple alignment of reference sequences registered in GenBank. The conserved region of the VP1 (ORF2) gene was chosen for BNoV, the membrane protein (M) gene for BToV, and the RNA polymerase (3D) gene for BKoV. We used hydrolysis probes, which were dual-labeled oligonucleotides bearing a 5′-fluorescent reporter dye (VIC, NED, FAM) and a 3′-nonfluorescent quencher (NFQ; Table 1).
Primers and hydrolysis probes used in our multiplex RT-qPCR testing for bovine norovirus (BNoV), bovine torovirus (BToV), and bovine kobuvirus (BKoV).

GenBank MH579349.
GenBank MN073058.
GenBank HQ650196.
Artificial templates and field samples
The 3 artificial templates (in vitro transcribed RNA) were prepared by inserting synthesized oligonucleotides into the pBHA vector cloning site and transcribing the RNA coding sequence (Bioneer). Oligonucleotides followed the sequence of the target gene to be amplified (BNoV: TTGGCCCTGATTTGAACCCTTATTTGGCCCACCTCCGTCGCATGTACAATGGGTGGACCGGATCCATG; BToV:
TCCAGCTTTGCATTGGTCGCCTATATGGCGCGGTTTGGAGAATTTTTGGCTTTTCTTGAGTCTTGCATCCC; BKoV:
GGGTGTGACCCTCCCATCCATCCTAGATACATCAAGGAGTTTTACGACAAACACACACCTCTCGTCGTGACTC). The target genes were sequenced for confirmation. The artificial templates were calculated for copy number and then diluted (RNA copy number: 108–101). Ct, assay linearity (correlation coefficient, R2), and reaction efficiency (efficiency, E) were measured to compare the performance of single and multiplex assays.
We obtained, by digital rectal sampling, 167 fecal samples from calves < 1-y-old with pasty-to-watery diarrhea in Chungbuk province, Korea, from 2016 to 2019. Nucleic acids were isolated from the samples (Patho Gene-spin plus extraction kit; iNtRON Biotechnology). Nucleic acid templates were stored at −80°C until use.
Reaction condition for singleplex and multiplex RT-qPCR assays
Reaction mixtures were prepared with 3 kinds of primers, probes, and artificial templates. Artificial nucleic acid templates for BNoV, BToV, and BKoV were amplified using synthesized primers and probe sets (Table 1; 2× 1-Step RT-qPCR master mix type-1 kit, Misogene). The assay was performed in a 20-µL reaction mixture containing 1× final concentration of master mix, 12 pmol of each primer, fluorogenic probe (BNoV, 4 pmol; BToV, 5 pmol; BKoV, 4 pmol), and 10 pg–100 ng of template. The final volume was adjusted using nuclease-free water. The concentration and volume of each component were optimized to maximize delta Rn (∆Rn) and minimize the Ct when amplifying the target gene. 10 Artificial and field sample templates were amplified (QuantStudio 5 real-time PCR system; Thermo Fisher) using the following conditions: reverse transcription at 50°C for 30 min, followed by incubation at 95°C for 15 min, 40 cycles of denaturation at 95°C for 20 s, and annealing and extension at 60°C for 1 min (total running time: ~2 h).
Analytical sensitivity and specificity of our multiplex RT-qPCR method
Sensitivity was measured by determining the limit of detection (LOD) of 10-fold serially diluted (108–100 copies/μL) and 25 and 50 copies/μL artificial templates. We used 23 replicates for each concentration (i.e., 11 × 23 reactions) of artificial template to confirm the LOD of the multiplex RT-qPCR assay. The LOD of the test was estimated as the lowest concentration that met a detection rate of 95%. 18 Furthermore, the LODs were compared with 10-fold serially diluted (100–10−7) viral templates obtained from BNoV-, BToV-, and BKoV-positive field samples between multiplex RT-qPCR and conventional RT-PCR assay for BNoV, BToV, and BKoV.19,20,23,27 Experiments were repeated 3 times; 8 concentrations (10-fold serially diluted field samples) were tested simultaneously in our multiplex RT-qPCR and conventional RT-PCR assays.
To determine specificity, we used prepared distilled water and RNA extracts from other clinically related causes of calf diarrhea (bovine rotavirus, bovine coronavirus, BVDV) and other viruses from the same families (porcine kobuvirus, bovine nebovirus). We assessed the detection rate of each virus that yielded true-negative results in our multiplex RT-qPCR assay. These non-bovine noro-, toro-, and kobuviruses were tested to confirm the exclusivity of the assay.
Simulation of single infection and coinfection by combining same concentrations of artificial templates
In vitro transcribed RNA of 1 (BNoV, BToV, BKoV), 2 (BNoV+BToV, BNoV+BKoV, BToV+BKoV), and 3 (BNoV+BToV+BKoV) pathogens at the same concentration were tested with our multiplex RT-qPCR assay. The concentrations used in the test were 1 × 107 and 1 × 102 copies/μL (LOD).
Evaluation of our multiplex RT-qPCR assay using clinical specimens
We collected 167 fecal samples from field animals infected with BNoV, BToV, or BKoV, or that were classified as negative for all 3 viruses. The extracted RNA from the field samples was analyzed using both our multiplex RT-qPCR and the conventional RT-PCR assays. If the sample was positive only by the conventional RT-PCR assay, the positive PCR product was confirmed by sequencing. In contrast, if the sample was positive only by our multiplex method, it was assessed using conventional RT-PCR (annealing temperature: 60°C), and the positive PCR product (~100 bp) was inserted into the pGEM-T Easy vector using TA cloning (Bioneer) and sequenced for confirmation.
Diagnostic sensitivity and specificity of our multiplex RT-qPCR assay
Diagnostic sensitivity and specificity were assessed by testing 167 fecal samples. Sensitivity was assessed using positive samples that were confirmed by sequencing.
Results
Validation of our multiplex RT-qPCR method
The performances of the multiplex and singleplex RT-qPCR assays were compared using standard curves (Fig. 1). Standard curves (Ct values vs. log10[RNA copies]) confirmed the linearity and efficiency of the singleplex and multiplex RT-qPCR assays in a wide range: 101–108 copies for the BNoV artificial template (singleplex: R2 = 0.998, E = 106%; multiplex: R2 = 0.998, E = 102%), 101–108 copies for the BToV artificial template (singleplex: R2 = 0.999; E = 110%; multiplex: R2 = 0.999, E = 108%), and 101–108 copies for the BKoV artificial template (singleplex: R2 = 0.999; E = 106%; multiplex: R2 = 0.999, E = 105%).
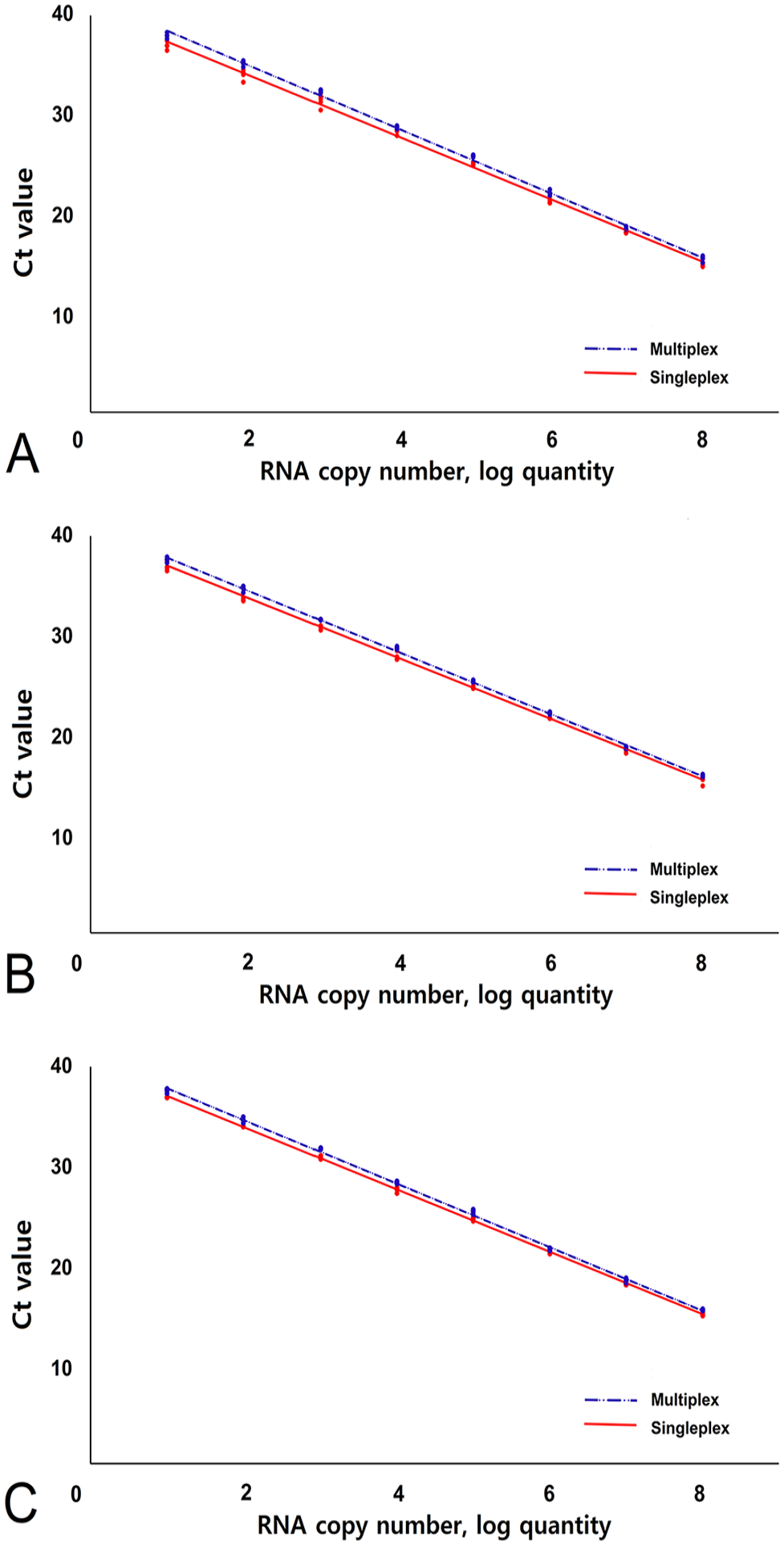
Standard curves of RT-qPCR for singleplex and multiplex assays for each artificial template (in vitro transcribed RNA) detection. RT-qPCR assay performance was measured by serial dilutions of artificial templates containing (
A cutoff for the positivity of the RT-qPCR assay was set at Ct = 38. The criteria were set by the LOD. The LOD was 1 × 102 copies/μL, with Ct = ~35 in multiplex RT-qPCR, and 1 × 101 copies/μL, with Ct = ~38 in some artificial template samples, but the detection rate was unqualified.
Analytical sensitivity and specificity of our multiplex RT-qPCR assay
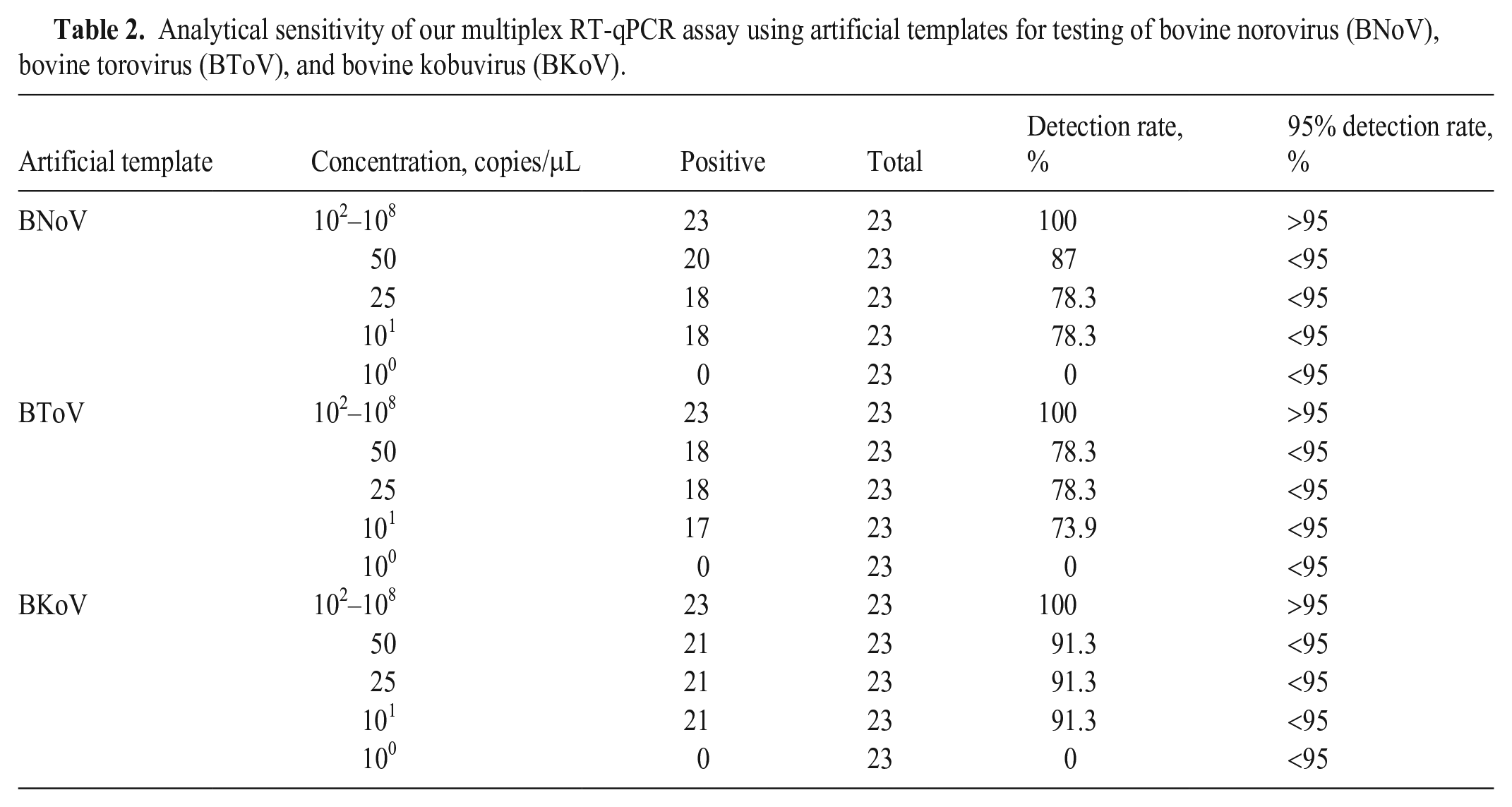
The LOD was the lowest concentration that yielded ≥ 95% detection of replicates. Thus, the LODs were 1 × 102 copies/μL for BNoV, BToV, and BKoV in our multiplex RT-qPCR assay using artificial templates (Table 2).
Analytical sensitivity of our multiplex RT-qPCR assay using artificial templates for testing of bovine norovirus (BNoV), bovine torovirus (BToV), and bovine kobuvirus (BKoV).
The LODs of the multiplex RT-qPCR and conventional RT-PCR assays using 10-fold serial dilution viral templates were 10−4 for BNoV and BToV and 10−6 for BKoV in our multiplex RT-qPCR assay, and 10−1 for BNoV, 10−4 for BToV, and 100 for BKoV in the conventional RT-PCR assay, respectively. Hence, the detection performance of our multiplex RT-qPCR assay was superior to that of the conventional RT-PCR assay.
No amplification curves were obtained with our multiplex RT-qPCR assay for distilled water, BNoV-, BToV-, and BKoV-negative field samples, or specific pathogen–positive field samples (porcine kobuvirus, bovine nebo-, rota-, and coronaviruses; BVDV).
Single infection and coinfection simulation assay
Artificial templates (BNoV, BToV, BKoV) at concentrations of 1 × 107 to 1 × 102 copies/μL were used for single (Fig. 2) and coinfection (Fig. 3) simulation experiments. Both high (1 × 107 copies/μL) and LOD (1 × 102 copies/μL) concentrations were reliably detected without delayed Ct values or undetectable pathogens.
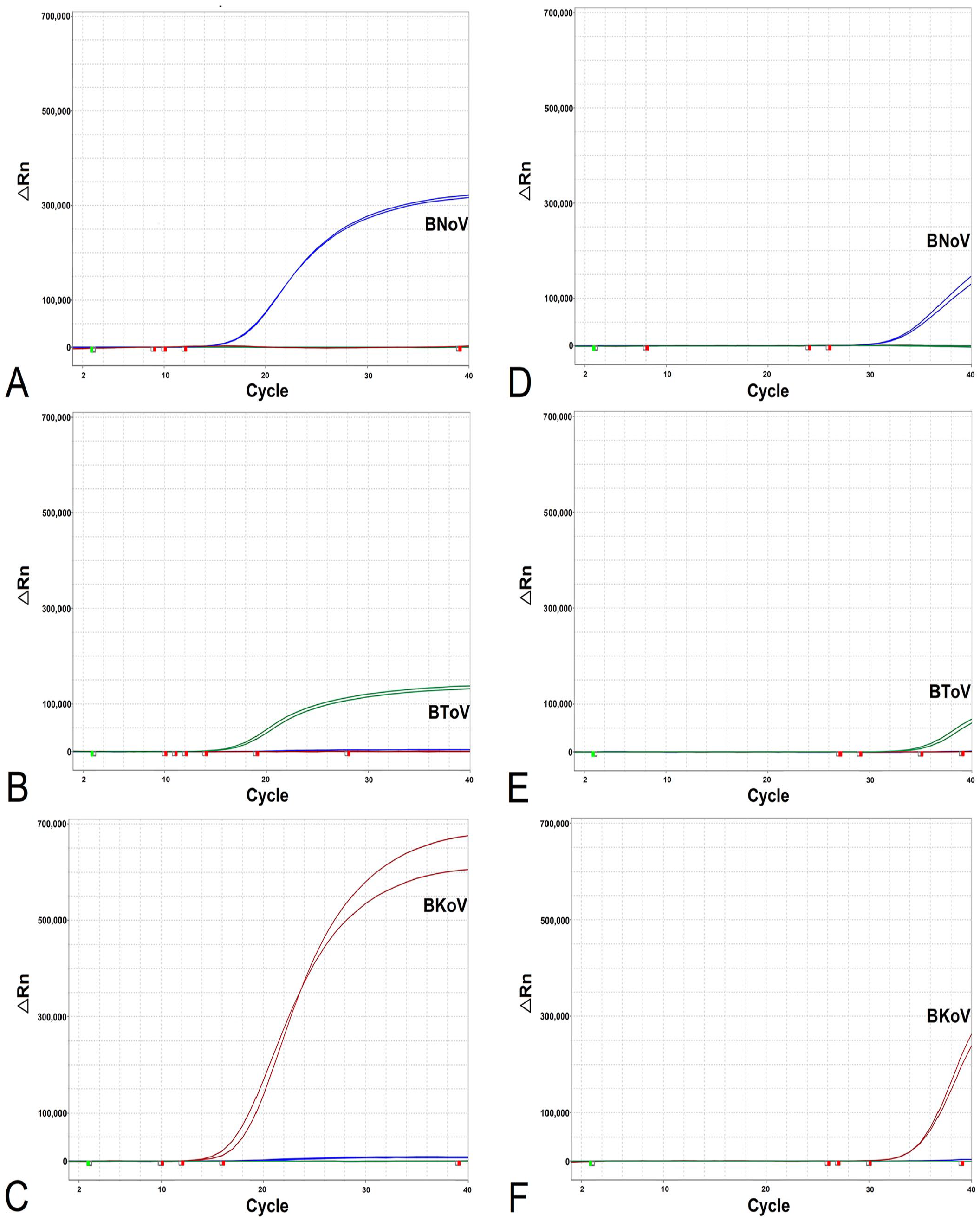
Single pathogen infection simulation experiments. Amplification curves (x-axis: cycle; y-axis: delta Rn) of (
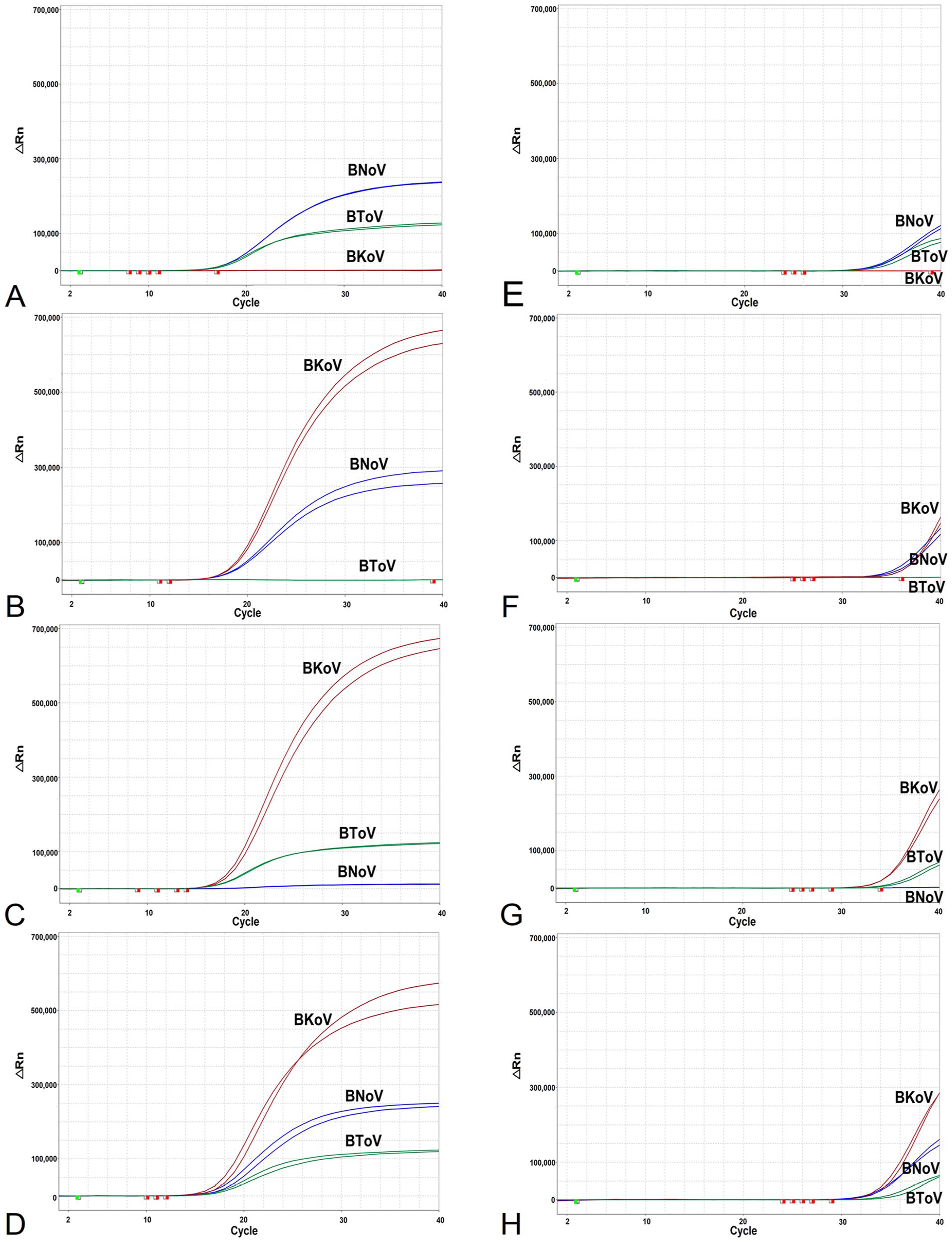
Coinfection simulation experiments with 2 and 3 pathogens. Amplification curves (x-axis: cycle; y-axis: delta Rn) of (
Detection of field samples using our multiplex RT-qPCR assay
Analysis of all 167 samples using our multiplex RT-qPCR and the conventional RT-PCR assays yielded identical results for 165, 167, and 164 samples for BNoV, BToV, and BKoV, respectively. Our multiplex RT-qPCR assay yielded 2 additional positives for BNoV and 3 additional positives for BKoV compared to the conventional RT-PCR assay. In the field samples in which ≥ 2 pathogens were detected, coinfections with BNoV and BToV were detected in 2 samples by both our multiplex RT-qPCR assay and the conventional RT-PCR assay. Coinfections with BNoV and BKoV were detected in 3 field samples in our multiplex RT-qPCR assay. The conventional RT-PCR assay failed to detect BKoV in 2 field samples. Coinfection with BToV and BKoV was detected in 1 field sample by our multiplex RT-qPCR assay; the conventional RT-PCR assay failed to detect BKoV in the field sample. No field samples were co-detected with BNoV, BToV, and BKoV (Table 3).
Comparison of our multiplex RT-qPCR assay with conventional RT-PCR assays for detection of bovine norovirus (BNoV), bovine torovirus (BToV), and bovine kobuvirus (BKoV) in field fecal samples.
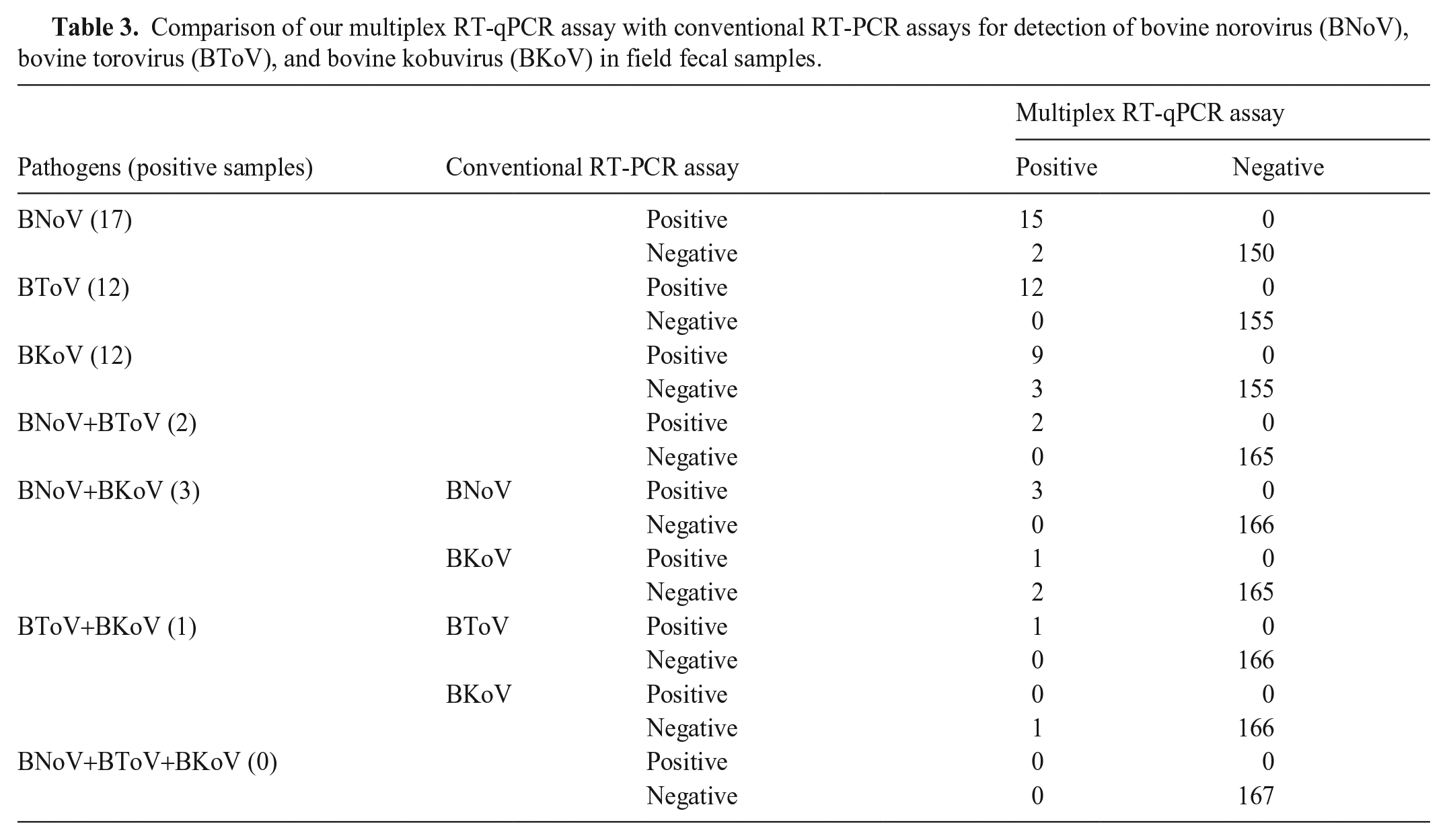
To determine whether each of the 5 field samples was a true- or false-positive, the 5 additional positive PCR products were inserted into a vector using TA cloning, and the nucleotide sequences were analyzed and compared. All 5 nucleotide sequences were identical to those of the corresponding reference strains registered in GenBank: MH579349 for norovirus; HQ650196 and JQ026109 for kobuvirus.
Diagnostic sensitivity and specificity of our multiplex RT-qPCR assay
Comparing the diagnostic sensitivity and specificity of the conventional RT-PCR and multiplex RT-qPCR assays with 39 samples confirmed positive through sequencing and 128 samples identified as negative, the conventional RT-PCR assay had a sensitivity of 87% (34 of 39) and specificity of 100% (128 of 128). The sensitivity of our multiplex RT-qPCR assay was 100% (39 of 39) with specificity of 100% (128 of 128). Thus, our multiplex RT-qPCR assay was more sensitive than the conventional RT-PCR assay.
Discussion
A multiplex RT-qPCR assay can detect multiple pathogens simultaneously; therefore, it is very economical and rapid because it decreases the required bench space, supplies, reagents, and labor to process samples. 8 Optimizing the amplification of the 3 target genes was necessary. Standard curves drawn after singleplex and multiplex RT-qPCR assays showed that R2 for each artificial template was ~1, and E included the recommended range of 90–110% in singleplex and 80–120% in multiplex. 3 Our multiplex RT-qPCR assay was unaffected by the presence of other primers and probes compared to the corresponding singleplex assays for BNoV, BToV, and BKoV.
To optimize our multiplex RT-qPCR assay, we evaluated methods for the detection of BNoV, BToV, and BKoV by measuring analytical sensitivity and specificity. The LOD of our multiplex RT-qPCR assay is low at 1 × 102 copies/μL for BNoV, BToV, and BKoV. In addition, comparing our multiplex RT-qPCR with conventional RT-PCR assays using field samples, detection limits were of the order of 10−4 for BNoV and BToV, and 10−6 for BKoV. BNoV and BToV had lower LODs in our multiplex RT-qPCR assay than in the conventional RT-PCR assay, and the LOD for BToV was the same for both methods. Therefore, our multiplex RT-qPCR assay could detect a low concentration of virus and was more sensitive than the conventional RT-PCR assays. Our multiplex RT-qPCR assay had high analytical specificity, with negative results in all negative field samples tested. Therefore, our primers and probes identified BNoV, BToV, and BKoV reliably.
In the simulation assay in which we manually created single and coinfection of BNoV, BToV, and BKoV, artificial templates could be stably detected without delayed Ct values or undetectable pathogens at both high (1 × 107 copies/μL) and LOD (1 × 102 copies/μL) concentrations. Our multiplex RT-qPCR assay detected infection by ≥ 2 pathogens.
The detection performance of our multiplex RT-qPCR assay in field samples was superior to that of the conventional RT-PCR assays. Among 167 samples tested, our multiplex RT-qPCR assay detected 2 more cases of BNoV and 3 more cases of BKoV than the conventional RT-PCR assays. In some samples, BKoV was negative in the conventional RT-PCR assay when coinfected with BNoV or BToV, but was detected by our multiplex RT-qPCR assay. The diagnostic sensitivity of our multiplex RT-qPCR assay was 100%, which was higher than that of the conventional RT-PCR assay (87%). The diagnostic specificity of our multiplex RT-qPCR assay was 100%, identical to that of the conventional RT-PCR method. Our multiplex RT-qPCR method is superior to the conventional RT-PCR assays given that it yielded faster, more sensitive, and more accurate detection. 18
Footnotes
Acknowledgements
We thank the Institute of Chungbuk Provincial Veterinary Service and Research (Cheongju, South Korea) for providing the environment and funding for the research.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Our study was conducted within the budget of the Institute of Chungbuk Provincial Veterinary Service and Research.