
Abstract
Droplet digital PCR (ddPCR) is a highly sensitive tool developed for the detection and quantification of short-sequence variants—a tool that offers unparalleled precision enabling measurement of smaller-fold changes. We describe here the use of ddPCR for the detection of Bovine leukemia virus (BLV) DNA provirus. Serum samples and whole blood from experimentally infected sheep and naturally infected cattle were analyzed through ddPCR to detect the BLV gp51 gene, and then compared with serologic and molecular tests. The ddPCR assay was significantly more accurate and sensitive than AGID, ELISA, nested PCR, and quantitative PCR. The limit of detection of ddPCR was 3.3 copies/µL, detecting positive experimentally infected sheep beginning at 6 d post-infection. The ddPCR methodology offers a promising tool for evaluating the BLV proviral load, particularly for the detection of low viral loads.
Bovine leukemia virus (BLV; Deltaretrovirus) is one of the most widespread livestock pathogens in dairy herds. It is estimated that a high proportion of herds are infected with BLV in North and South America.3,34,42 Approximately 40% of BLV-infected cattle will develop persistent lymphocytosis within a few years of infection; <5% of infected cattle develop lymphosarcoma.3,13 The high prevalence of BLV infection is the result of the common occurrence of infected cattle that remain subclinical, a condition referred to as aleukemic.4,13 With little effort to control the spread of BLV, the virus is widely prevalent in many places.
BLV infection can impact innate and adaptive immune system cells and alter the proper functioning of uninfected cells. 12 BLV infection may also induce changes in the complex balance of cytokine expression, cell proliferation, and programmed cell death in both T- and B-lymphocytes, which is critical for immune competence and effective response to infectious challenge. 10 Thus, a decreased immune response to immunization may be expected in seropositive animals. Indeed, low-magnitude serologic responses to a commercial foot-and-mouth disease vaccine 36 and a J5 Escherichia coli vaccine 10 have been observed. Dairy cows infected with BLV have altered immune function associated with decreased milk production and shortened lifespan.2,42
Although cattle and water buffaloes are the natural hosts of BLV, other species can be infected experimentally, with different clinical, hematologic, and immunologic manifestations. Sheep are not naturally infected with BLV, 52 but can become infected using blood from BLV-infected cattle, infectious BLV molecular clones, or tumor-derived cells. 35 Experimentally infected sheep can be used as a model to test for BLV to avoid false negatives in cattle tests. Sheep in particular exhibit interesting biological features for evaluating BLV pathogenesis as well as for diagnostic purposes. BLV-infected sheep develop tumors typical of the disease and display signs of lymphosarcoma 2.5 y after BLV infection, which is much faster than in cattle. 7 The fetal lamb kidney (FLK) cell line persistently infected with BLV (FLK-BLV) is one of the long-term cultures used most commonly for the production of virus. 35
Laboratory techniques must detect subclinical infections accurately because animals infected subclinically represent 90% of BLV infections, especially those with recent infections or with low proviral load (PVL). The World Organisation for Animal Health (OIE) recommends the use of ELISA and agar gel immunodiffusion (AGID) tests for antibody detection, and nested PCR (nPCR) for agent identification; real-time PCR (rtPCR) is also now incorporated. 49 The rtPCR has been implemented widely for clinical viral load testing, but relatively poor precision has hindered its usefulness. 15
Classic BLV eradication programs rely on the correct identification and segregation or elimination of BLV-infected animals.41,42 However, serologic tests may fail to detect BLV-specific antibodies in recently infected animals (false negatives) or may detect maternal antibodies in calves borne to BLV-positive cows.9,29 To date, at least 20 countries have eradicated BLV successfully through control programs, yet BLV remains highly prevalent (up to 90%) in many regions of the world, including Eastern Europe, South America, and several Asian countries. 34 The primary management practices used to prevent new infections are early identification and segregation or culling of infected cattle.3,25 Furthermore, in certain physiologic states, such as the peripartum, the natural decrease of specific anti-BLV antibodies may produce false-negative ELISA results from day −20 to 60. 38 Whole blood PVLs may be a good indicator of disease progression in the field, 21 making PVL useful for disease control programs, given that it has been shown that animals with low PVLs are less effective in viral transmission20,26 and that they can be kept near negative herds.
As a feasible approach to control the virus, selection of cattle carrying the BoLA-DRB3*0902 allele has been proposed, given that this allele is strongly associated with a BLV infection profile or the low PVL phenotype. 20 Therefore, the inclusion of highly sensitive screening assays might increase the efficiency of an eradication program, especially for herds with low within-herd prevalence.
The precise quantification of BLV in subclinical animals in countries in which the prevalence of infection is medium or high has been suggested as a key strategy for viral control in dairy cattle.14,42 However, more evidence is necessary to demonstrate the effectiveness in the control of transmission from the segregation of the herd at high and low PVLs. It may be that this strategy has not been applied on a large scale because methodologies that allow viral quantification in a simple, precise, and reproducible way and in different matrices (blood or milk) have not been developed. Until recently, quantitative PCR (qPCR) was the only option, which has limitations such as the need for a reference gene, and its low efficiency in the presence of matrices with inhibitors. The inadequacy of qPCR was demonstrated in a comparison of the performance of 6 different qPCR assays, demonstrating wide discordance in the ability to detect and quantify BLV infections. 19 These limitations are partially or totally resolved with droplet digital PCR (ddPCR), suggesting that ddPCR would be an excellent alternative for BLV control programs.
ddPCR is a highly sensitive tool developed for the detection and quantification of a range of microorganisms, 48 including other deltaretroviruses, such as the human T-cell lymphotropic virus (HTLV1; Primate T-lymphotropic virus 1). 51 Digital PCR advantages include DNA quantification without the need for calibration curves, thereby allowing for exact values to be obtained easily.39,43 Digital PCR can measure smaller copy number variations than qPCR 47 and can determine the absolute quantification of samples with higher precision and reduced PCR bias, reducing influencing factors and making results more reliable and reproducible.5,33,51 Digital PCR allows small percentage differences to be detected and variants to be quantified, a significant advantage compared to existing tools (AGID, ELISA, nPCR, rtPCR) for the detection and control of BLV.
Our objective was to standardize a ddPCR or digital PCR (last-generation PCR) assay for the detection and quantification of the proviral form of BLV in subclinical animals and to compare the results with those obtained using reference techniques.
Materials and methods
Controls
As a positive control, we used DNA extracted from an FLK-BLV cell line. These cells were cultured in RPMI 1640 medium (Corning) supplemented with 5% inactivated fetal bovine serum. A gp51 glycoprotein recombinant plasmid (vector Ptop-Blunt-V2; Macrogen) was used as an additional positive control. The expected copy number was calculated according to the molecular weight of the plasmid and the DNA concentration. As a negative control, genomic DNA (gDNA) from species unrelated to the natural BLV host was used (ovine, equine, canine species); ultra-pure water was used as a no-template control (NTC) to avoid false negatives.
Experimental infection of sheep with BLV
Experimental BLV infection was carried out in a sheep model.8,35 Four adult female Corriedale sheep (~2-y-old) were given an IM injection of 1 mL of infection solution (10 µL of whole blood from a BLV+ naturally infected cow, in 1 mL of PBS), and 2 other sheep were used as uninfected controls kept in the same conditions. The sheep were kept on a sheep farm in southern Uruguay in a natural pasture field under typical feeding conditions, with ad libitum access to water. Blood samples (15 mL) were taken with a 19-G needle from the jugular vein of each animal into tubes with and without EDTA to obtain DNA and serum on 0, 2, 6, 9, 13, 21, 30, and 60 d post-infection (dpi). The experimental protocol was approved by the Ethical Committee on the Use of Animals (CEUA-FVET-Exp. 111900-000572-20).
Cattle naturally infected with BLV
Holstein heifers (n = 34), belonging to a cattle breeding farm in Uruguay, were analyzed for BLV antibodies every 3 mo for 12 mo by ELISA (Fig. 1) and nPCR at time 0 and 12 mo to obtain BLV-positive and -negative groups. Blood samples (10 mL), with and without anticoagulant to obtain DNA and sera, were taken from all animals by coccygeal venipuncture and stored at −20°C until processed. Antibodies were measured every ~2 mo (5 times in total); nPCR was performed at 0 d and at 12 mo in all animals. The experimental protocol was approved by the Ethical Committee on the Use of Animals (CEUA-FVET-Exp. 111100-000068-15).
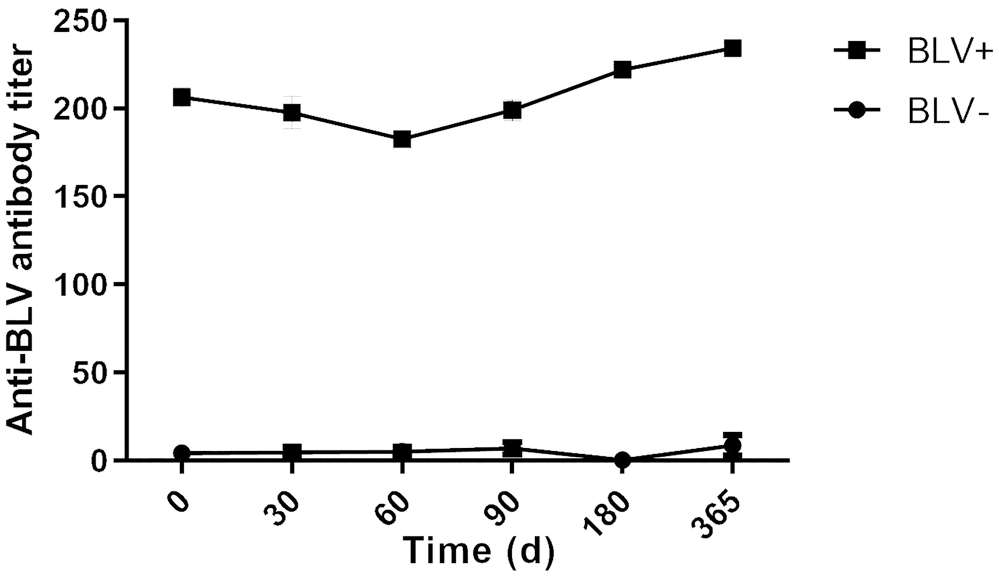
Bovine leukemia virus (BLV) mean antibody titer determined by ELISA for heifers >1-y-old analyzed by droplet digital PCR to determine BLV− or BLV+ status.
Determination of anti-BLV antibodies by AGID and ELISA
AGID was performed with a commercial kit (FCV-UNLP-Argentina) following the manufacturer’s instructions and the recommendations of the OIE. 49 Antigen and test sera were combined (35 µL each) and plated on 1% agarose gel plates. The precipitation bands (positive results) were visualized with indirect light 72 h post-incubation at room temperature in a humid chamber.
Anti-BLV antibodies were detected in individual sera with a blocking ELISA (P02140-10; Idexx) according to the manufacturer’s instructions and the recommendations of the OIE. 49 A 2× dilution of 50 µL of serum was used, and results were read at an optical density of 450 nm in a microplate spectrophotometer.
DNA extraction and digestion
DNA extraction from whole blood samples was performed using a phenol-chloroform protocol. 22 Isolated DNA was quantified with a UV-Vis spectrophotometer (NanoDrop; Thermo Fisher) and quality evaluated on agarose gels. After extraction, the DNA was digested with BamHI (Bioron) at 37°C for 15 min and the enzyme inactivated by heating at 65°C for 20 min. 44
Nested PCR
The nPCR targeted a region of 444 bp on the env BLV gene (the gp51 coding gene). Two rounds of amplification used the following primers: F1: 5′-TCTGTGCCAAGTCTCCCAGATA-3′; R1: 5′-AACAACAACCTCTGGGAA-3′; F2: 5′-CCCACAAGGGCGGCGCCGGTTT-3′; R2: 5′-GCGAGGCCGGGTCCAGAGCTGG-3′. The 25-µL reactions contained 12.5 µL of 2× MangoMix (Bioline), 1 µL (10 μM) of each primer, and 50 ng of DNA template. The result of the amplification was visualized in 1.5% agarose gels in TAE 1× buffer (40 mM Tris, 20 mM acetic acid, 1 mM EDTA, pH 8.3), stained with GoodView nucleic acid stain (SBS Genetech).11,34
Real-time PCR
BLV proviral DNA quantification was performed using a SYBR green fluorescence assay (7500 real time PCR system; Applied Biosystems). The qPCR was performed following a modified protocol,32,40 using the following primers: F: 5′-CCTCAATTCCCTTTAAACTA-3′ and R: 5′-GTACCGGGAAGACTGGATTA-3′. The sensitivity of these BLV SYBR qPCR assays was described as 2 copies per reaction. 32 The reaction was performed in a volume of 25 µL containing 12.5 µL of 2× Fast start SYBR green kit (Roche), 1 µL (10 µM) of each primer, and 100 ng of DNA template. The thermocycler profile included a pre-denaturation of 2 min at 50°C, then denaturation for 10 min at 95°C, followed by 40 cycles of 15 s at 95°C, 15 s at 55°C, and a final extension of 60 s at 60°C. The plasmid pBLV1 containing the BLV pol gene was used as a standard. Ten-fold dilutions of this standard were made: 5 × 106 to 5 × 100 copies/μL. Positive results were classified in high and low PVLs, with a cutoff of 2,000 copies/µg of DNA. 14
Primer and probe design for ddPCR
The primers and probe were designed using Primer3web v.4.1.0 (https://primer3.ut.ee/),23,24,46 physicochemical parameters were calculated by NetPrime (PREMIER Biosoft International; http://www.premierbiosoft.com/netprimer/), and specificity and spurious pairing were determined with NCBI Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome). 50
No artifacts were observed on the qPCR SYBR green melting curve. Artifacts were investigated using 2 experiments in rtPCR, the first without DNA, varying the concentration of the forward and reverse primers in the final concentrations of 0.5 mM, 0.25 mM, 0.125 mM, 0.062 mM, and 0.031 mM (Table 1). The second experiment used concentrations of 0.35 mM, 0.5 mM, 0.75 mM, 1.25 mM, and DNA from 39 samples to determine the influence of target DNA on the formation of artifacts.
Experimental design for primer concentration optimization measured as cycle threshold.
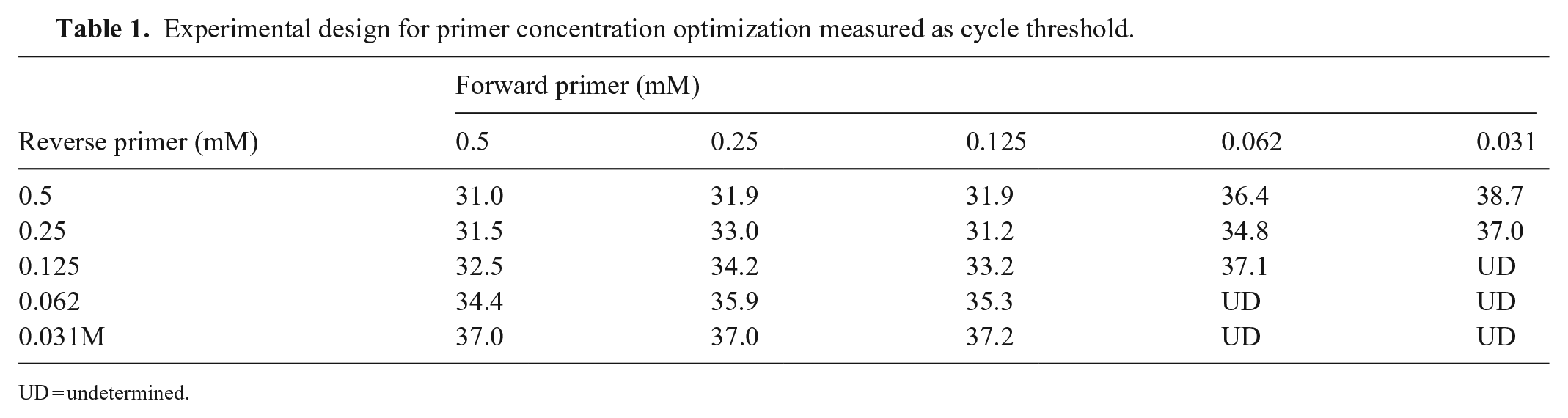
UD = undetermined.
Droplet digital PCR for BLV quantification
ddPCR was performed (QX200 droplet digital PCR system; Bio-Rad) per the manufacturer’s protocol. The probe and primer sequences were designed to amplify a partial segment of the BLV env gene. The primer sequences were F: 5′-CAGTGACTGGGTTCCCTCTGTC-3′ and R: 5′-AGGGCGAGRCCGGGTCCAGAG-3′; the probe was HEX-5′-CCCTCCCTGGGCTCCCGARA-3′-BHQ1. Briefly, in a typical 20-µL assay, 1 µL of DNA sample was mixed with 1 µL of each primer (10 μM), 0.5 µL of probe (10 μM), and 2 × Supermix emulsified with oil (Bio-Rad); the droplets were transferred to a 96-well plate (Eppendorf). The PCR assay was performed in a thermocycler (C1000 touch cycler; Bio-Rad) with the following parameters: initial denaturation of 10 min at 95°C, then 40 cycles of 30 s at 94°C, and 10 min at 58°C, with final deactivation of the enzyme for 10 min at 98°C. The presence of fluorescent droplets determined the number of resulting positive events that were analyzed in the software (QuantaSoft v.1.7.4; Bio-Rad), using dot charts. The BLV PVL was determined as copies/µL of bovine gDNA. Each sample was run in duplicate, and results were averaged.
Sensitivity and specificity of ddPCR
In order to initially evaluate the sensitivity (how well the test could identify true positives) and specificity (how well the test could identify true negatives) of this proposed ddPCR method, we analyzed a cell line persistently infected with BLV (FLK) and a recombinant plasmid containing BLV as positive samples. We also tested the ability of the technique to detect the virus both in highly pure samples (plasmids) and in samples of cells infected with the virus.30,31 In addition, DNA from complete genomes of unrelated animal species was used as negative control.
To gauge the accuracy of ddPCR as a qualitative method, the results of a set of samples were compared with other standard technics. BLV-infected sheep were defined as true positives; uninfected sheep were used as true negatives. Serum and DNA samples from both groups (uninfected and infected animals) were analyzed by AGID, ELISA, nPCR, and ddPCR. Sensitivity (true positive rate) and specificity (true negative rate) were calculated (v.9.0.1.1; MedCalc). We also compared results of herd samples obtained by qPCR and ddPCR; mean copy number and the Pearson correlation coefficient for time were calculated.
The digested DNA was compared to undigested DNA using ddPCR with sample dilutions of 1:10, 1:100, 1:1,000, and 1:10,000 of single samples (F3542) in a one-step reaction with 15 min of digestion by BamHI.
Validation of quantitative ddPCR method
Five 10-fold dilutions of the linearized recombinant plasmid containing the gp51 sequence (vector Ptop-Blunt-V2), in triplicate, and 2 NTC were quantified by ddPCR to validate the quantitative method according to ISO/IEC 17025:2017 (https://www.iso.org/standard/66912.html). Linear regression was used to estimate an analytical curve characterized by the slope (analytical sensitivity), intercept, and confirmed by R2. Assuming confidence of 95%, the standard error at the intercept (SE) permitted calculation of the limit of detection (LOD = 3.3 × [SD of intercept/slope]) and limit of quantification (LOQ = 10[SD of intercept/slope]). For selectivity analysis, DNA extracted from blood from ovine, equine, and canine species and from uninfected bovine or ovine sera were tested. Other selective analysis of the primers designed for ddPCR was performed in silico using Primer-BLAST (consulting non-redundant [nr] databank).
Results
Primer and probe design
The primers and probe were designed to amplify a small region of the BLV env gene gp51. Three sets of primers and probes were generated, and the set with the best physicochemical parameters was chosen. For this set, with a 162-bp amplicon size, the self-dimer ΔG was −2.84 Kcal/mol and −6.91 Kcal/mol for the forward and reverse primers, respectively, and the cross dimer ΔG was −5.35 Kcal/mol. The melting temperature (Tm) was at least 60°C for each primer and 70°C for the probe. The theoretical Tm was tested empirically, indicating that 60°C was the ideal temperature. Our search on Primer-BLAST returned 8,670 hits; a maximum of 2 variable loci were detected in the primer, which was compensated for by incorporation of degenerate primers with satisfactory hybridization to both primers and probe.
Empirical tests showed that qPCR with 0.1–0.5 mM primers, in the absence of DNA template, generated nonspecific amplification (Ct = 32 and 30.9, respectively). However, in the presence of target DNA or nonspecific DNA template, this amplification disappeared. The reactions carried out with the probe did not show nonspecific amplification.
Standardization and validation of quantitative ddPCR
The assay performed well on the ddPCR platform, showing a LOD in the scatterplots of event number (droplets) versus fluorescence amplitude (Fig. 2). The QuantaSoft software provides autoanalysis as well as manual thresholding tools that allow correct designation of positive and negative droplet populations. Positive drops were determined by fluorescence intensity using QuantaSoft software. Only drops that exceeded the threshold value were counted as positive. The linear regression of 17 points (5 plasmid dilutions in triplicate and 2 NTC) generated an analytical curve with linear correlation R2 = 0.9952, sensitivity (slope) of 17,780, and intercept of −0.2306. Assuming a 95% confidence level, we could calculate a LOD of 3 copies/µL of reaction volume and a LOQ of 11 copies/µL of reaction volume. We used 1 µL in a typical reaction. In a qualitative analysis, animals with a ddPCR result of ≥3 copies/µL of reaction were considered positive.
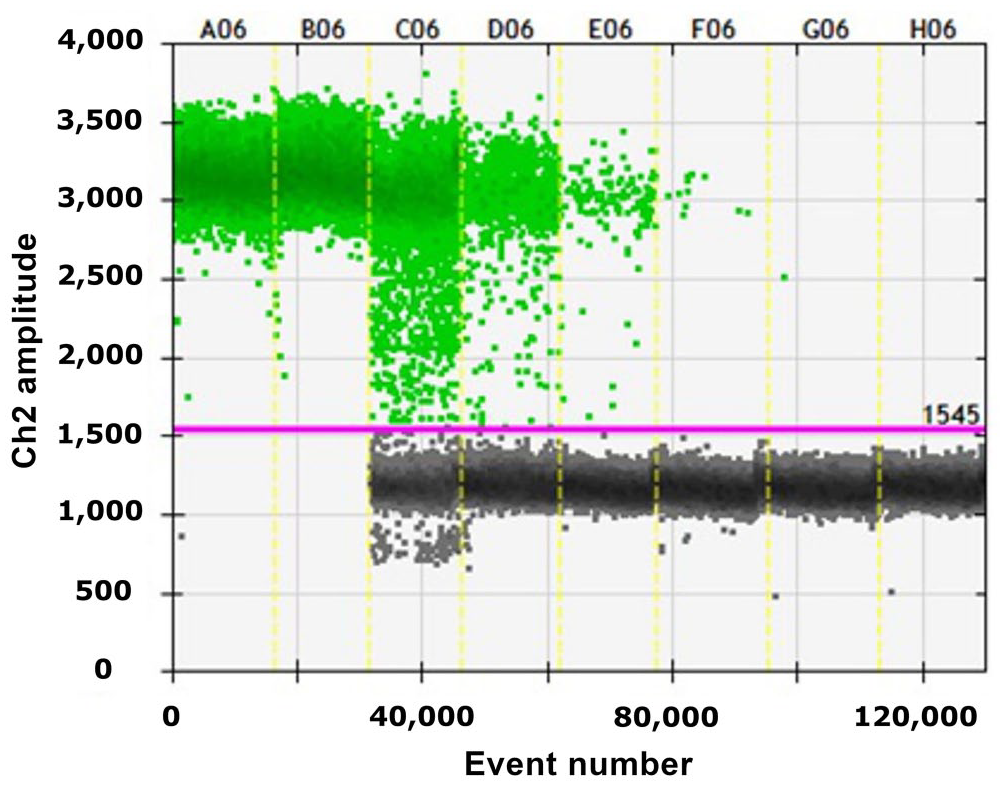
Output from droplet digital PCR of plasmid dilutions containing the bovine leukemia virus gene gp51 from 0.02 ng/µL to 0.02 × 10−6 ng/µL (A06–G06) and NTC (H06). Gray = negative events; green = positive events.
The selectivity analysis for ddPCR showed no amplification when whole blood gDNA from a sheep, a horse, and a dog, and BLV-uninfected sheep were tested. The in-silico analysis of the primers with Primer-BLAST found similarities only to BLV (909 of 932 matches) in queries made to nr databank, considering virus (taxid: 10239) or retrovirus (taxid: 11632) subsets, respectively.
Sheep infected experimentally with BLV
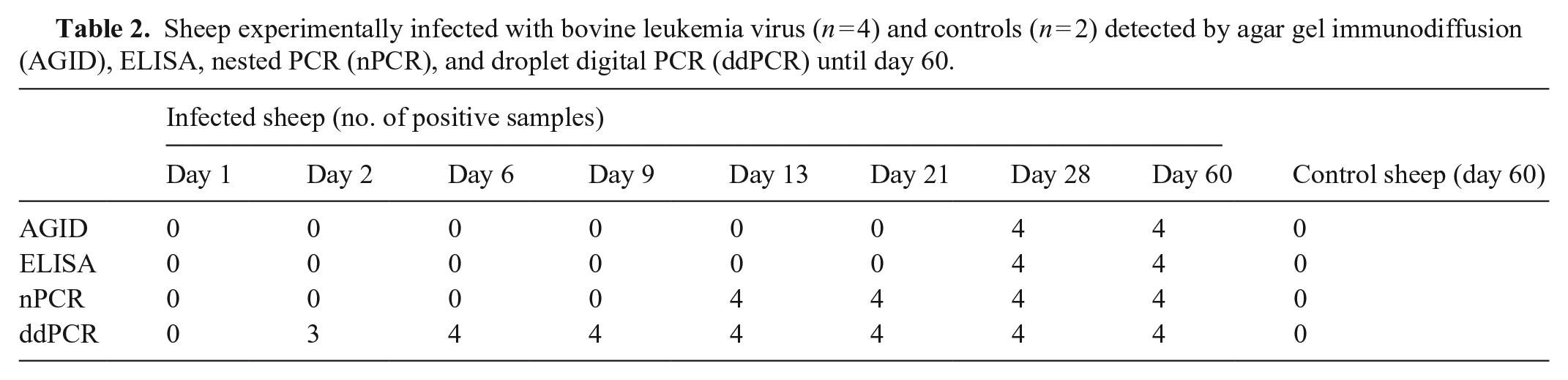
From 13 dpi, the 4 BLV-inoculated sheep were positive by nPCR. Specific antibodies were detected on day 28 in all infected sheep (4 of 4) by AGID and ELISA. Uninfected sheep remained negative by nPCR and serologic tests throughout the experiment (Table 2). On the other hand, ddPCR detected BLV from 2 dpi in 3 of 4 BLV-positive sheep and at 6 dpi in all animals. Control sheep remained negative throughout the study by ddPCR (Table 2; Fig. 3).
Sheep experimentally infected with bovine leukemia virus (n = 4) and controls (n = 2) detected by agar gel immunodiffusion (AGID), ELISA, nested PCR (nPCR), and droplet digital PCR (ddPCR) until day 60.
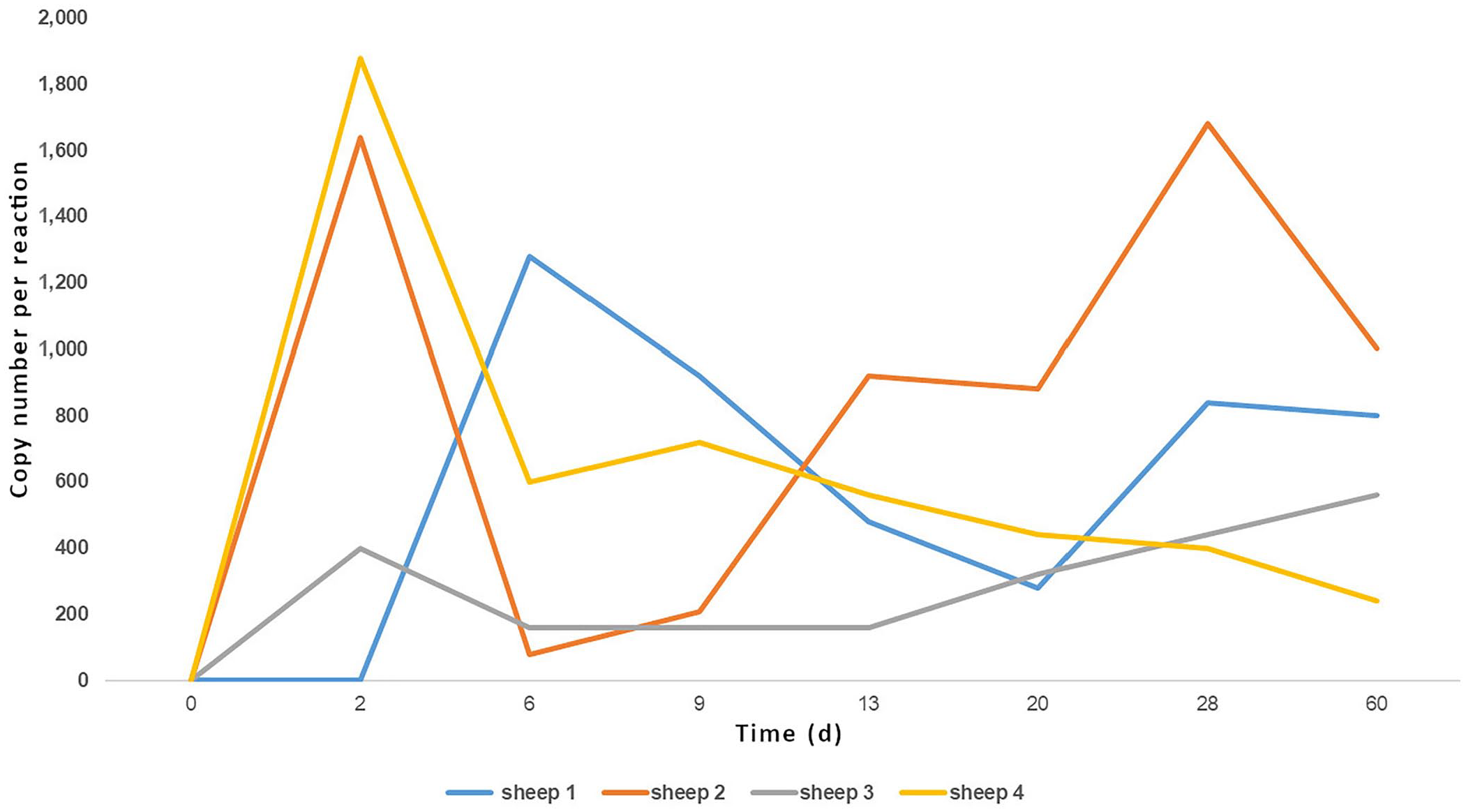
Number of copies of gp51 from bovine leukemia virus–infected sheep quantified by droplet digital PCR.
Cattle infected naturally with BLV
Of the 34 animals analyzed, 20 were positive (BLV+) and 14 remained negative (BLV−) for 12 mo by ELISA and nPCR. The samples from both groups were analyzed by qPCR and ddPCR. Of the animals determined to be negative by ELISA and nPCR, 100% were negative by qPCR, whereas 35% were positive by ddPCR (Table 3). The ddPCR-positive samples were sent for sequencing, and the presence of the BLV gp51 gene was confirmed (data not shown). Of the positive samples that were analyzed twice (0 and 365 d), the mean PVL measured by qPCR at time 0 was 8,266 copies/µg DNA, and at 365 d was 16,510 copies/µg DNA. For ddPCR, the count at time 0 was 29,325 copies/µg DNA, and at 365 d was 26,611 copies/µg DNA (Fig. 4). When classified by viral load (high and low load), the tests coincided in 50% of the samples.
Molecular detection of bovine leukemia virus in ELISA-negative heifers after 1-y observation in an infected herd. Results of real-time PCR (rtPCR) and droplet digital PCR (ddPCR) in copies/µL.
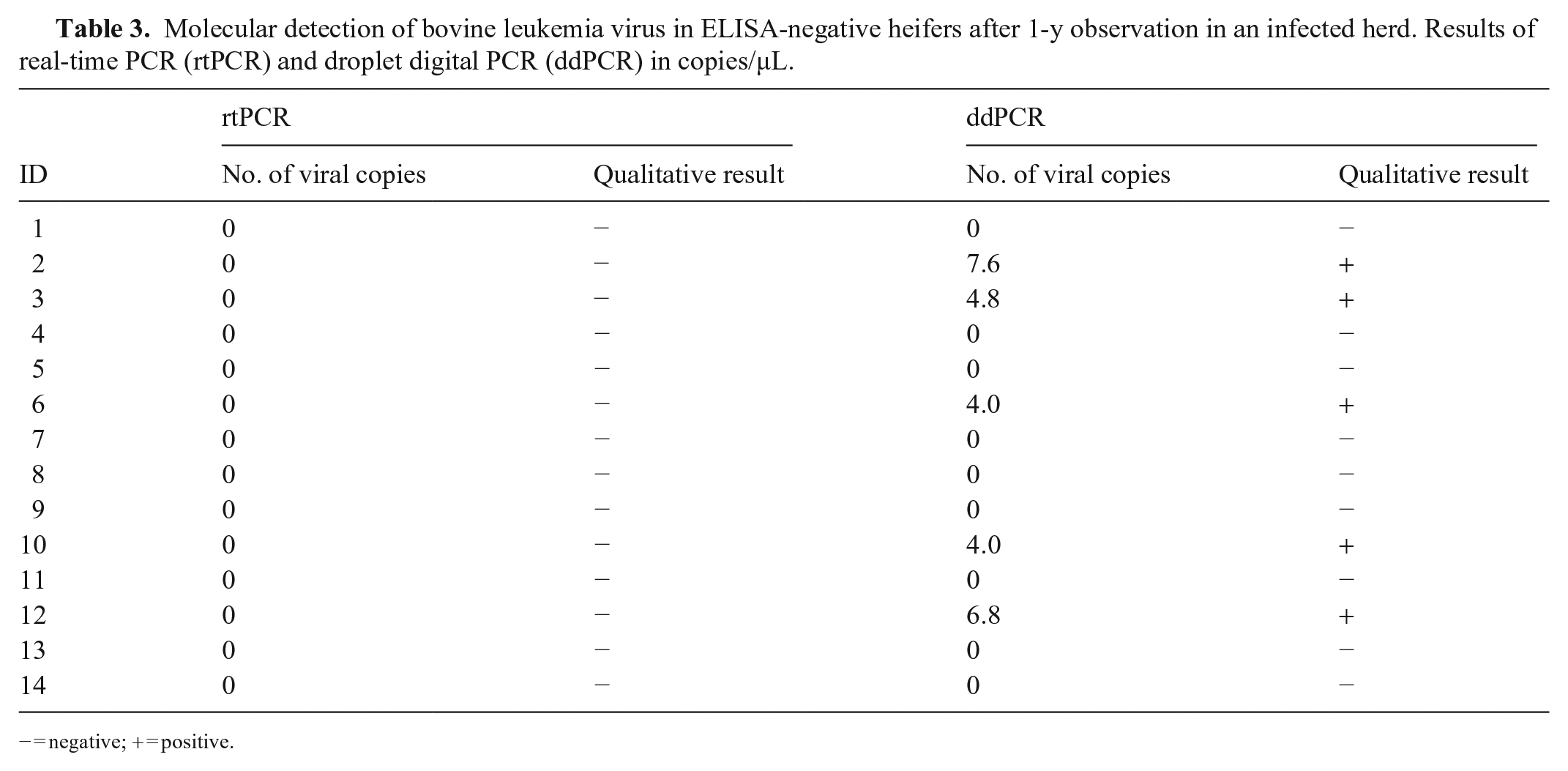
− = negative; + = positive.
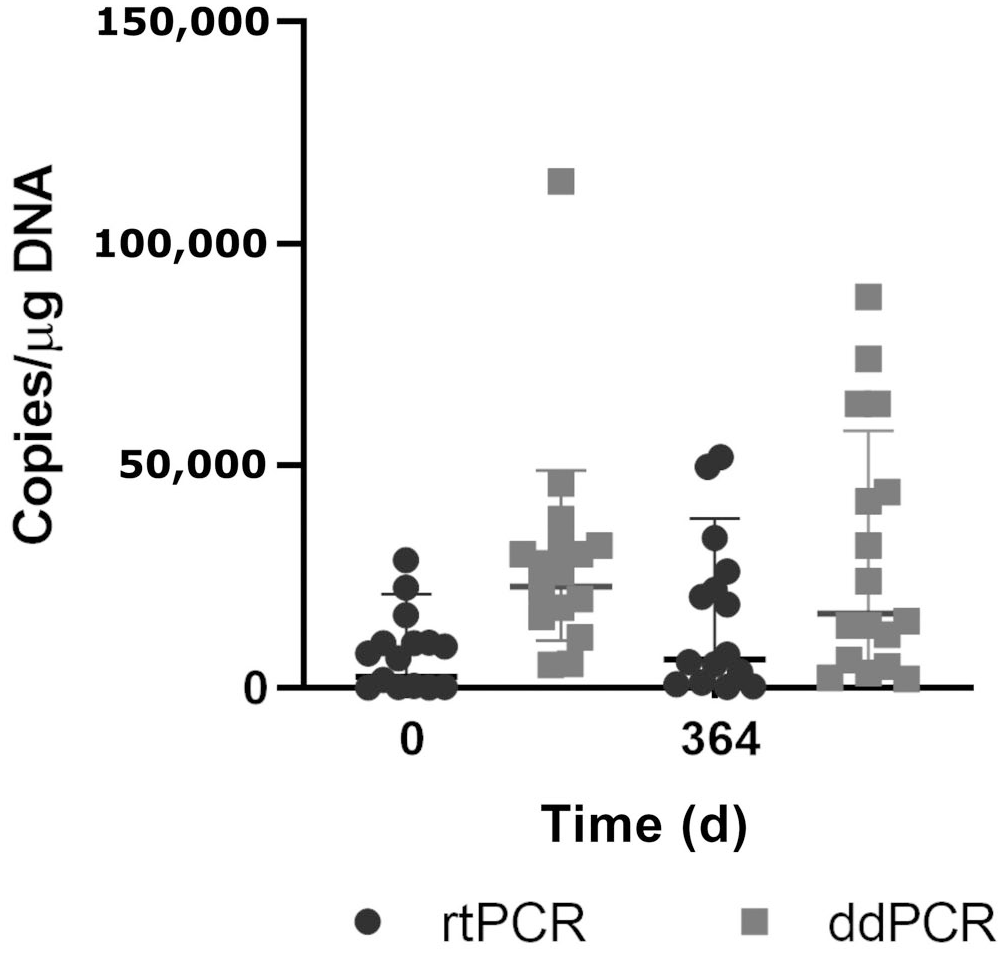
Quantification of bovine leukemia virus (BLV) proviral load by droplet digital PCR (ddPCR) and real-time PCR (rtPCR) of BLV-positive Holstein heifers over 1 y. Central bar = mean; whiskers = 1 SD.
Sensitivity specificity
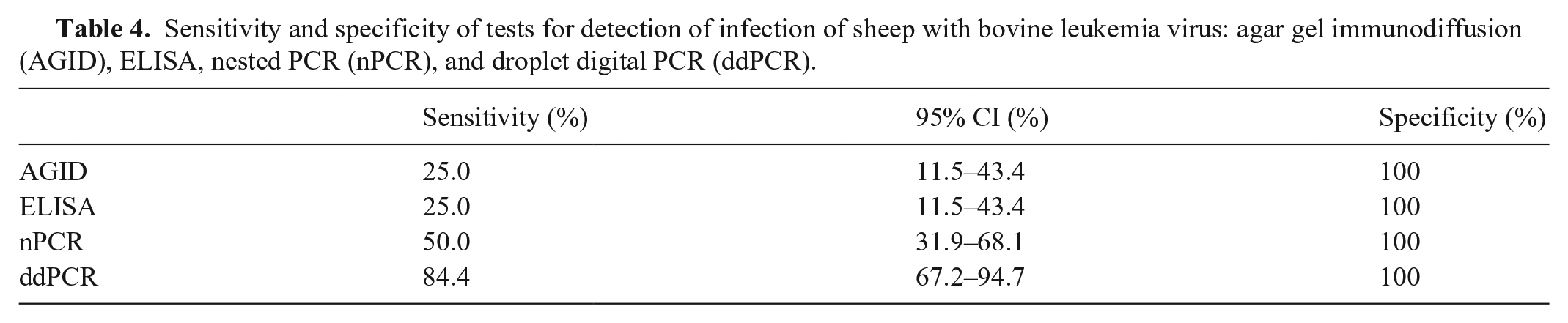
The proposed ddPCR method was able to detect BLV in the FLK cell line and recombinant plasmid. Otherwise, no amplification was obtained from uninfected animal gDNA. The provirus was not detected in the control sheep (not experimentally infected) over the test period, demonstrating 100% specificity of the ddPCR in true-negative samples. The ddPCR was able to detect BLV from 2 dpi, whereas nPCR positive detection was at 13 dpi (Tables 2 and 4). The possibility that these were false-positive results was ruled out by sequencing digital PCR products and confirming 100% similarity with the BLV gp51 gene deposited at GenBank using BLAST (data not shown).
Sensitivity and specificity of tests for detection of infection of sheep with bovine leukemia virus: agar gel immunodiffusion (AGID), ELISA, nested PCR (nPCR), and droplet digital PCR (ddPCR).
Digestion of gDNA could potentially increase the sensitivity of detection or BLV in positive samples. The results analysis found 40% more copies/µL in the digested sample than the nondigested sample, but results were irregular at 1:1,000 and 1:10,000.
Discussion
Quantification of PVL using ddPCR has been established for human retroviruses, wherein the PVL in HTLV1 infections was used as a marker of disease progression. 45 Based on the similarities in pathogenesis between HTLV and BLV, we optimized ddPCR for BLV in peripheral blood of naturally infected cattle and experimentally infected sheep. The resulting ddPCR was highly sensitive and specific for BLV from various infected animals, making it suitable for use in early detection, to clarify inconclusive samples analyzed by qPCR, as well as for PVL quantification. Droplet digital PCR has not been optimized previously for a provirus of animal origin, to our knowledge, and this technique could be applied to other viruses of veterinary interest.
The primary challenge of the ddPCR approach is avoiding accidental subquantification of the proviral gene, which can be avoided through the complete digestion of the host genome with restriction enzymes, with the caveat that the digest does not affect the provirus assay. With this precaution, we found ~40% more copies of BLV after digesting the samples. Although this strategy significantly increases the sensitivity of the test,1,16 there may still be droplets with more than one viral target.
Our ddPCR assay was able to detect positive sheep from 2 dpi, whereas nPCR, the OIE reference method, detected positive sheep from 13 dpi. Although not a replacement for nPCR, the greater sensitivity of ddPCR has an advantage for the detection of BLV in the earliest days of infection, when the proviral load is very low We did not compare the sensitivity of ddPCR to qPCR; however, it would be expected that the sensitivity of qPCR is similar to that of nPCR.19,32
Using control sheep not experimentally infected, the provirus was not detected over the test period in these animals, demonstrating 100% specificity of the ddPCR in true-negative samples. The use of DNA from cattle (the natural host of BLV) as truly negative samples is not feasible given that the prevalence of BLV in dairy cattle in Uruguay is 80–90%, increasing the possibility of false negatives. We analyzed samples from 14 heifers that were seronegative for BLV by ELISA during 1 y, and 5 tested positive for BLV by digital PCR. To confirm that these were true positives and rule out the possibility of cross-contamination in the DNA samples, these 5 animals were tested in duplicate, and results were confirmed by sequencing (data not shown). The LOD of this technique is 3 copies/µL, an improvement compared to 5 copies/µL detectable by qPCR of blood samples, 32 which may explain the 5 new positive samples. It may be that these animals carried an undetectable permanently low PVL. Based on these results, we recommend the cautious use of samples from negative cattle diagnosed by the currently available tests, when comparing detection tools or immune responses to BLV.
In addition to nucleic acid detection, ddPCR is a platform designed to provide high precision for quantification of target molecules because it carries out direct quantification without requiring a calibration curve.17,45 In contrast, the rtPCR assays used widely in diagnostic laboratories around the world show significant variability in the measurement of the BLV proviral DNA copy number among laboratories, even when qPCR is performed by experienced staff.28,32,40 We found that PVLs were not concordant between qPCR and ddPCR. In general terms, ddPCR detected a significantly higher number of virus copies than qPCR, which can be explained by the higher sensitivity and resistance to inhibitors of ddPCR.17,27,37 This can be an important advantage in BLV control if we intend to use easily accessible samples such as milk (bulk tank or individual), but that have significantly less provirus and more PCR inhibitors than blood samples. Other authors have shown that the ddPCR assay was more tolerant to sodium dodecyl sulfate and heparin than the qPCR assay. This phenomenon is a result of reaction partitioning; the occurrence of individual reactions in each droplet diminishes the impact of inhibitors on PCR amplification by retaining the positive signal even when moderate PCR inhibition occurs in a droplet. 6 Resistance to PCR inhibitors is a crucial advantage for future development of ddPCR in milk samples for BLV control.
We detected antibodies by ELISA and AGID ~1 mo post-infection in sheep and 15 d after positive nPCR detection. Similar findings have been described after the experimental infection of steers, but proviral DNA was detected via rtPCR at 24 dpi (almost 10 d after the ddPCR) and antibodies by ELISA at 36 dpi. 18 When we analyzed samples from cattle, we found animals with low PVLs by ddPCR that were seronegative by ELISA during all tests (12 mo). This is likely because, with a low amount of circulating virus, the immune response is limited, and antibody titers are not detected by conventional tools.
The high resistance to inhibitors, independence from the need for standard curves, and inter- and intra-laboratory reproducibility of ddPCR make the method attractive. The correlation with qPCR in determining PVL was not conclusive and should be reviewed in future research.
Footnotes
Acknowledgements
We thank the staff of ‘Campo de Recría La Cruz’ (Florida, Uruguay) for supporting field activities, and veterinarians Drs. Gustavo Sacco and Agustin Furtado. We also thank Dr. Scott Brandl for his constructive suggestions and English editing, and Dr. Martín Breijo for the follow-up in the writing of the manuscript.
Declaration of conflicting interests
The authors declared no conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed the receipt of the following financial support for research, authorship and/or publication of this article: Our project was funded by the Agencia Nacional de Investigación e Innovación (FSSA_X_2014_1_105283-ANII).