
Abstract
Canine parvovirus 2 (CPV-2) and feline panleukopenia virus (FPLV) often cause acute enteric disease in their hosts. A simple, rapid, and effective method for the on-site detection of these viruses would be useful. We used a denaturation bubble-mediated strand exchange amplification (SEA) method to successfully detect CPV-2 and FPLV in fecal samples. SEA could detect as little as 3.6 pg/μL of CPV-2 and 6.6 pg/μL of FPLV genomic DNA following a 40-min incubation at an isothermal temperature of 61°C. Unlike PCR, SEA does not require complicated equipment, and positive samples produce a color change that can be visualized by the naked eye. Additionally, SEA is simpler than PCR because no extraction is needed, and heating of the fecal sample at 98°C can be performed with a heating block or water bath. This rapid and effective nucleic acid detection platform could be used as a point-of-care test for the detection of CPV-2 and FPLV.
Introduction
Canine parvovirus 2 (CPV-2; Carnivore protoparvovirus 1), which appears to share a common ancestor with feline panleukopenia virus (FPLV; Carnivore protoparvovirus 1), was first isolated from the feces of dogs suffering from enteritis in the late 1970s.1,2 After years of mutation and recombination of the major capsid gene (VP2), the original CPV-2 has evolved into 3 CPV-2 antigenic variants (CPV-2a, -2b, and -2c),3,16,24 which can cause disease in both dogs and cats. 4 CPV-2 and FPLV are the leading enteric pathogens in domestic and wild dogs, especially in unvaccinated puppies, and cats, respectively.5,10
Viral enteritis caused by CPV-2 or FPLV is a major threat to pet dogs and cats. CPV-2 and FPLV infections occur mainly through the fecal-oral route and can cause enteric disease, including fever, diarrhea, vomiting, and other clinical signs, sometimes resulting in a fulminant epidemic. 12 Early and rapid diagnosis is crucial for disease management and to prevent the spread of CPV-2 and FPLV. A method that can rapidly detect CPV-2 and FPLV infection in resource-limited settings would be valuable. Some traditional assays, such as ELISA, 13 hemagglutination assays, 17 and immunochromatography tests, 19 have low specificity and sensitivity. With the development of molecular technology, TaqMan real-time PCR (rtPCR) and gel-based PCR have been used to detect CPV-2 with various degrees of specificity and sensitivity.6,9 In addition, isothermal amplification methods such as loop-mediated isothermal amplification (LAMP),11,18 recombinase polymerase amplification, 26 helicase-dependent amplification, 25 polymerase spiral reaction, 8 and insulated isothermal PCR 28 have been used for CPV-2 detection. Compared with these nucleic acid amplification methods, denaturation bubble-mediated strand exchange amplification (SEA) requires only a pair of common primers and a Bst DNA polymerase. 21 Since the establishment of SEA in 2016, SEA has been used not only to detect various pathogens, such as Escherichia coli, 22 Listeria monocytogenes, 29 Mycoplasma pneumonia, 23 and Bursaphelenchus xylophilus, 14 but also to address the problem of meat adulteration.15,27
We evaluated the SEA method for the detection of CPV-2 and FPLV in fecal samples. This approach was expected to achieve rapid, visual detection without relying on sophisticated instruments, and hence to be suitable for resource-limited settings and point-of-care testing.
Materials and methods
Sample preparation
Stool samples were obtained from Qingdao Agricultural University (China). Clinical samples were collected from 35 dogs and 18 cats with classical signs of CPV-2 and FPLV infection (anorexia, vomiting, diarrhea, and fever), respectively. The CPV-2 strain isolated from F81 cells was used as a positive control. The FPLV positive control was strain MK301396, which had been isolated from the fecal sample of a cat with enteritis. The negative sample used in the specificity test was not infected with CPV or FPLV, which had been confirmed by PCR.
CPV-2 and FPLV viral DNA, as well as F81 cell DNA, were extracted (High Pure viral nucleic acid kit; Roche) according to the manufacturer’s instructions. Complementary DNA (cDNA) samples of canine coronavirus (CCV), canine distemper virus (CDV), feline coronavirus (FCoV), and feline calicivirus (FCV) were obtained from Qingdao Agricultural University. Viral genome DNA concentrations were measured (NanoDrop spectrophotometer ND-1000; Thermo Fisher Scientific). CPV-2 genomic DNA (gDNA; 3.6 × 102 pg/μL) was used to optimize the SEA reaction temperature. Detection of CPV-2 gDNA (3.6 × 103 pg/μL) and FPLV gDNA (6.6 × 103 pg/μL) was attempted to assess the feasibility of SEA. The sensitivity of the SEA assay was estimated using serial 10-fold dilutions of gDNA of CPV-2 (3.6 × 104 pg/μL to 0.36 pg/μL) and gDNA of FPLV (6.6 × 104 pg/μL to 0.66 pg/μL). The specificity of SEA was tested using gDNA from CPV-2, FPLV, F81 cells, and the negative sample, and cDNA from CDV, CCV, FCoV, and FCV.
To release the DNA targets and then test using SEA and LAMP, CPV-2 and FPLV viral DNA were extracted by heating the clinical fecal samples from 53 sick dogs and cats. The supernatants from fecal lysate and CPV-2–infected cell culture medium were heated at 98°C for 10 min in a simple heating block or water bath to release the DNA targets, and the resulting supernatants were added directly to the reaction system to initiate the SEA reaction.
Primer design
The VP2 gene of CPV-2 and FPLV is highly conserved. We downloaded 45 complete VP2 gene sequences (Suppl. Table 1), including different antigenic variants, from the NCBI dataset and aligned them using ClustalW in MEGA 5.0 (https://www.megasoftware.net/). Then, the most conserved region of the VP2 gene (GenBank accession KF149982.1) was selected as the SEA and LAMP target for designing specific primers. Primers were designed using Primer Premier 5.0 (Premier Biosoft) and assessed using NUPACK (http://www.nupack.org/) and DINAMelt Web Server (http://unafold.rna.albany.edu/?q=DINAMelt; Table 1; Suppl. Table 2). All primers were synthesized and purified by Sangon Biotech.
Primers used in the SEA assay and the LAMP assay of CPV-2 and FPLV.
BIP = backward inner primer; B3 = outer backward primer; FIP = forward inner primer; F3 = outer forward primer; LAMP = loop-mediated isothermal amplification; LB = loop backward primer; LF = loop forward primer; P1 = forward primer of SEA (strand exchange amplification); P2 = reverse primer of SEA.
SEA amplification reaction
SEA fluorescence detection and colorimetric kits, as well as LAMP fluorescent detection kits, were purchased from Qingdao Navid Biotechnology. Other reagents were purchased as follows: 6× DNA loading buffer, 20 bp DNA marker, and DNase/RNase-free distilled water (Sangon Biotech); methylene diacrylamide and acrylamide (MilliporeSigma); and tetramethylethylenediamine (TEMED) and ammonium persulfate (APS; Beijing Solarbio Science and Technology). The SEA fluorescent assay was performed in a 10-µL reaction mixture containing 1.0 μL of viral DNA, 1.0 µL of 10× isothermal buffer, 0.8 µL of dNTPs (10 mM), 0.25 µL of 20× EvaGreen, 0.2 µL of Bst 2.0 WarmStart DNA polymerase (8 U/µL), 0.25 µL of PEG-200, 0.1 µL of ET SSB (extreme thermostable single-stranded DNA binding protein; 500 µg/mL), 1.5 μL of each of the P1 (forward primer of SEA) and P2 (reverse primer of SEA) primers (10−5 M), and 3.5 μL of DNase/RNase-free distilled water. The reaction mixture was incubated at 61°C for 60 min in a heating block (Pad3-100C; Coyote Biotech), and SEA amplifications were monitored (CFX Connect real-time PCR system; Bio-Rad) at 1-min intervals. Then, 5 μL of amplified product from the SEA assay underwent 12.5% polyacrylamide gel electrophoresis (PAGE), and gel images were recorded (ChampGel5000 system; SageCreation Science; Fig. 1). Alternatively, amplification products could be confirmed by the pre-addition of a pH-sensitive dye, and the color change could be directly observed by the naked eye.
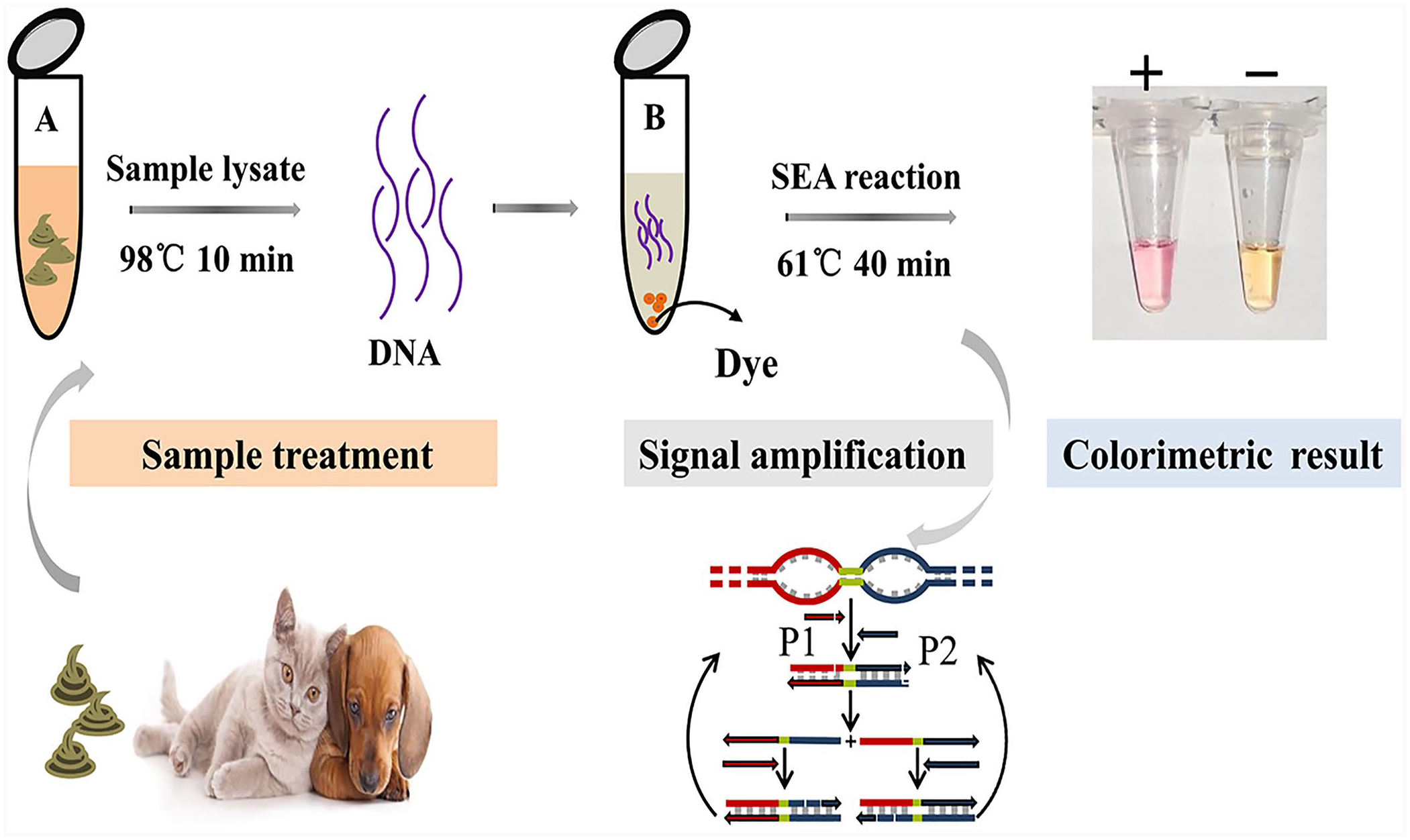
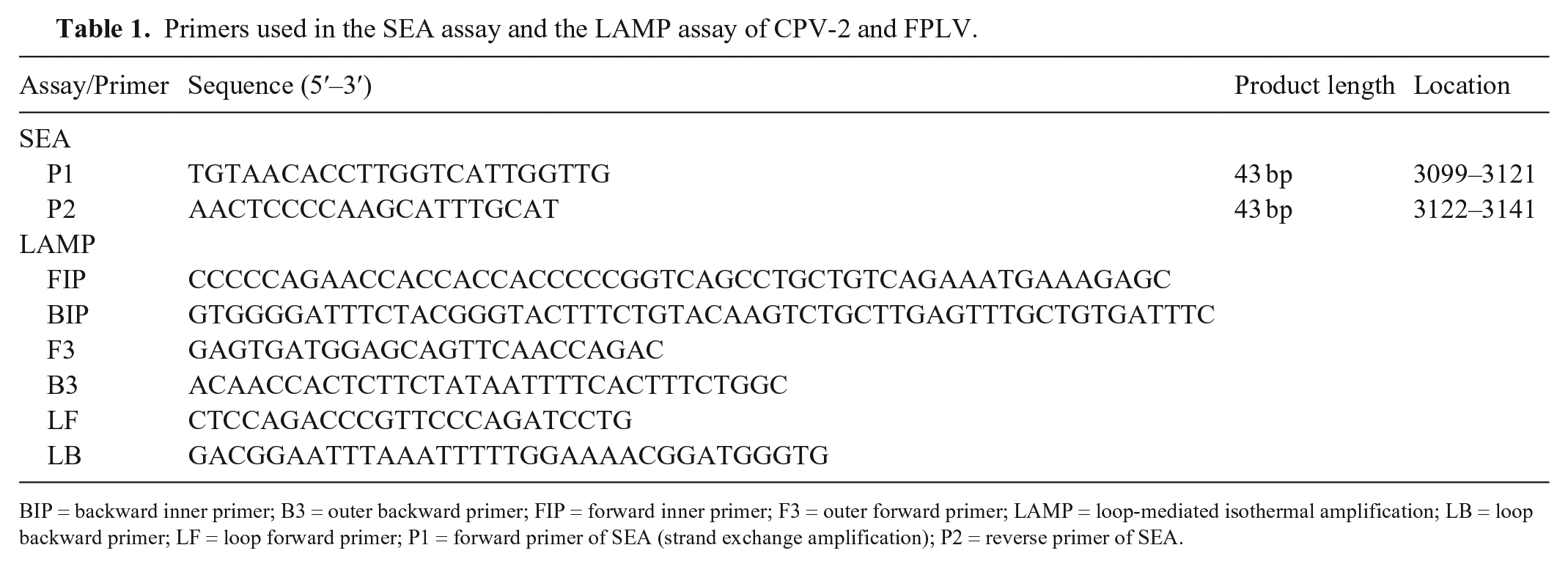
Schematic illustration of CPV-2 and FPLV detection by the isothermal SEA method with rapid sample processing. Tube A contains lysate in 0.9% normal saline. Tube B is the SEA reaction system. The SEA reaction result is determined by direct visualization. P1 = forward primer of SEA; P2 = reverse primer of SEA.
LAMP reaction
LAMP reactions were performed in a 10-µL volume consisting of 1.0 µL of template, 1.0 µL of 10× isothermal buffer, 1.6 μL of dNTPs (10 mM), 0.25 µL of 20× EvaGreen, 0.2 µL of Bst 2.0 DNA polymerase (8 U/µL), 0.2 μL of MgSO4 (100 mM), 0.2 μL of each of the F3 (outer forward primer) and B3 (outer backward primer; 10−5 M) primers, 1.6 μL of each of the FIP (forward inner primer) and BIP (backward inner primer; 10−5 M) primers, 0.8 μL of each of the LF (loop forward primer) and LB (loop backward primer; 10−5 M) primers, and 0.55 µL of DNase/RNase-free distilled water. The mixture was incubated at 63°C for 60 min (CFX Connect real-time PCR system; Bio-Rad).
PCR amplification for full-length VP2
Genomic DNA was amplified by PCR using Taq DNA polymerase with 2 pairs of primers (Suppl. Table 2). The PCR system contained of 2 μL of viral DNA, 0.1 μL of Taq DNA polymerase (2.5 U/μL), 2 μL of 10× Taq buffer, 0.6 μL of dNTP mixture (10 mM), 1 μL of MgCl2 (25 mM), 0.4 μL of each of the 2 pairs of primers (10−5 M), and 13.5 μL of DNase/RNase-free distilled water. PCR was performed using the following condition: 95°C for 30 s, 30 cycles of amplification (95°C for 30 s, 54°C for 30 s, and 72°C for 1.5 min), and a final extension at 72°C for 10 min. All amplified products were sequenced (Sangon Biotech), and VP2 full-length sequences (1,755 bp) were assembled (SeqMan software; DNASTAR). The nucleotide sequences were translated into amino acids and analyzed using DNAMAN (Lynnon Biosoft). The Taq DNA polymerase (2.5 U/μL), 10× Taq buffer, dNTP mixture (10 mM), and MgCl2 (25 mM) were purchased from Tiangen Biotech.
Results
Optimization of the SEA reaction temperature
Fluorescence curve 4 (Suppl. Fig. 1) had the shortest threshold time (Tt) compared with other temperatures, indicating that 61°C was the optimal SEA reaction temperature to detect CPV-2. Therefore, SEA reactions, including both fluorescence and colorimetric reactions, were conducted at 61°C.
Feasibility of SEA to detect CPV-2 and FPLV
Compared with the no-template control (NTC), gDNA of CPV-2 and FPLV extracted from both positive control samples had an obvious fluorescence signal after amplification, with short Tt values of 18 and 20 min, respectively (Fig. 2). Hence, SEA could effectively detect CPV-2 and FPLV, which was consistent with the electrophoresis results. The corresponding 43-bp amplification product appeared in the native PAGE (Fig. 2 inset, bottom). Notably, the colorimetric assay was conducted using only a heating block. The positive reaction changed from orange to purple-red at ~40 min, when the fluorescence curve tended to plateau, but the NTC reaction remained the original orange color (Fig. 2 inset, top). Furthermore, the repeatability of SEA was estimated (Suppl. Fig. 2), and good stability was observed for CPV-2 identification.
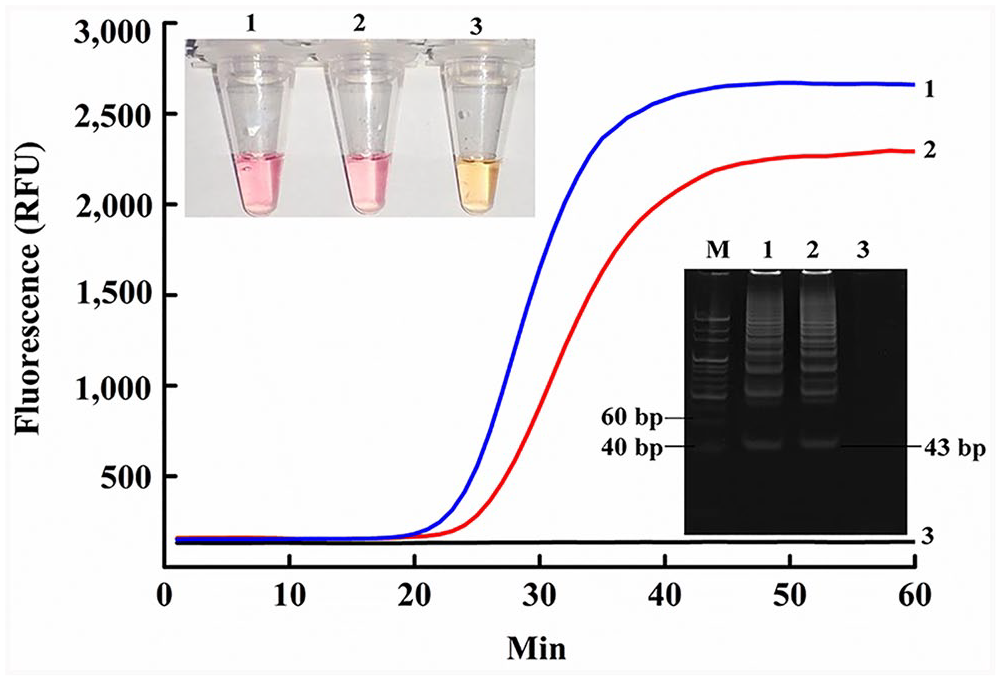
CPV-2 (line 1 in the fluorescence graph) and FPLV (line 2 in the fluorescence graph) gDNA were used to assess the feasibility of SEA. Relative fluorescence unit (RFU) in the y-axis is the fluorescence signal intensity after SEA amplification. Inset bottom right, amplified products (43 bp) in PAGE electrophoresis: lane M = 20-bp marker; 1 = CPV-2 gDNA; 2 = FPLV gDNA; 3 = no-template control (NTC). Inset top left are the SEA colorimetric reactions: 1 = CPV-2 gDNA; 2 = FPLV gDNA; 3 = NTC.
Sensitivity of SEA to detect CPV-2 and FPLV
The SEA method could detect as little as 3.6 pg/μL of CPV-2 DNA from fecal samples based on real-time fluorescent signals within 40 min (Fig. 3A). A linear relationship was observed between the Tt and negative logarithm (lg) of the CPV-2 gDNA concentration (Fig. 3B), generating a corresponding correlation coefficient (R2) of 0.9908 and the linear correlation equation Tt = 21.19 − 5.23 (lgCDNA). Similarly, the sensitivity of SEA for FPLV gDNA detection was 6.6 pg/μL (Fig. 4A), and a linear relationship was observed between the Tt and negative logarithm (lg) of the FPLV gDNA concentration (Fig. 4B), generating a corresponding R2 value of 0.9907 and the linear correlation equation Tt = 19.56 − 4.77 (lgCDNA). Although the SEA method and other traditional detection methods commonly used for CPV-2 detection are less sensitive than rtPCR, the analytical sensitivity obtained in our work was sufficient for the detection of both CPV-2 and FPLV, and the SEA method showed good linearity in detection of both CPV-2 and FPLV gDNA.
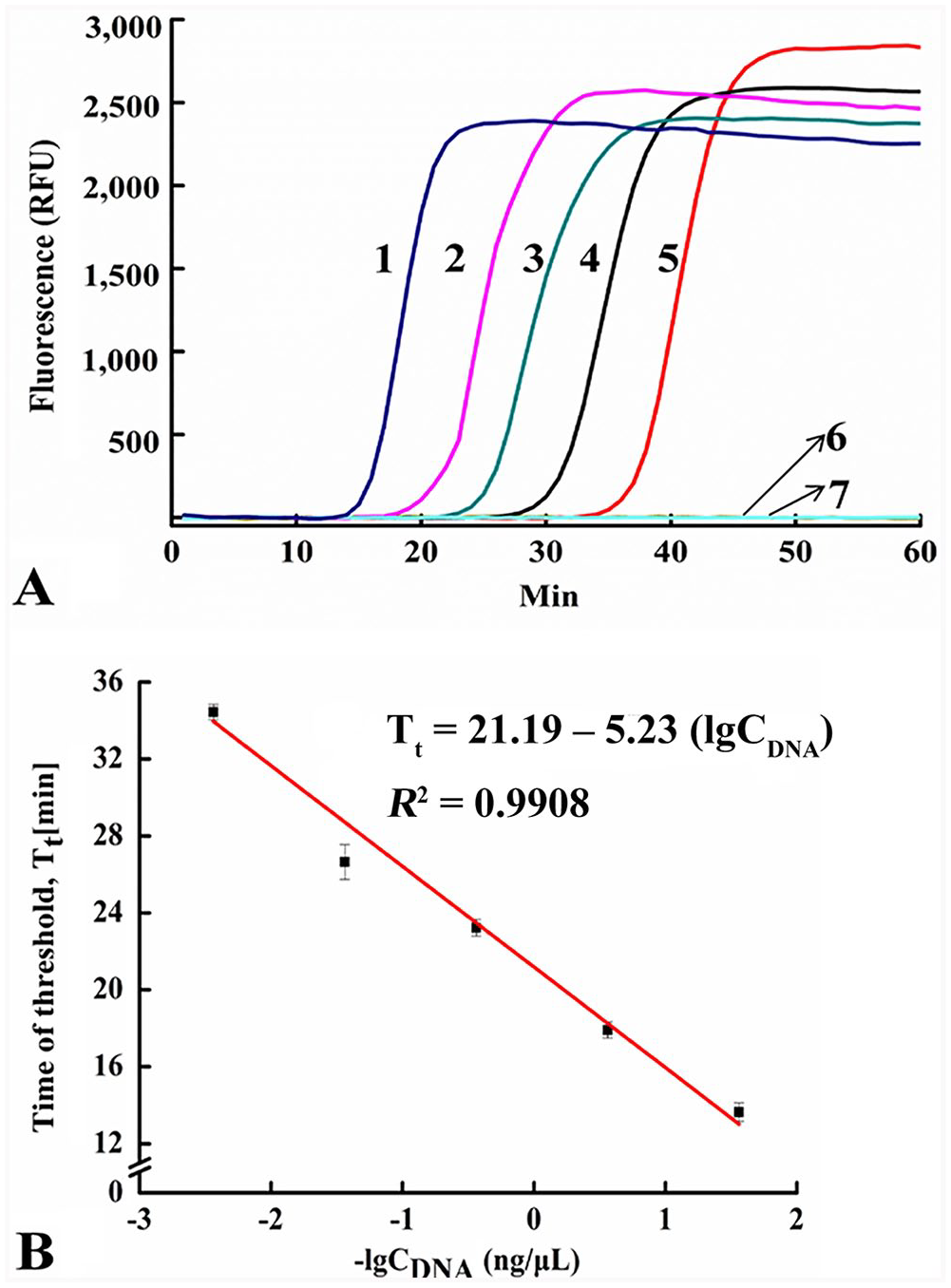
The sensitivity of the SEA method to detect CPV-2 gDNA.
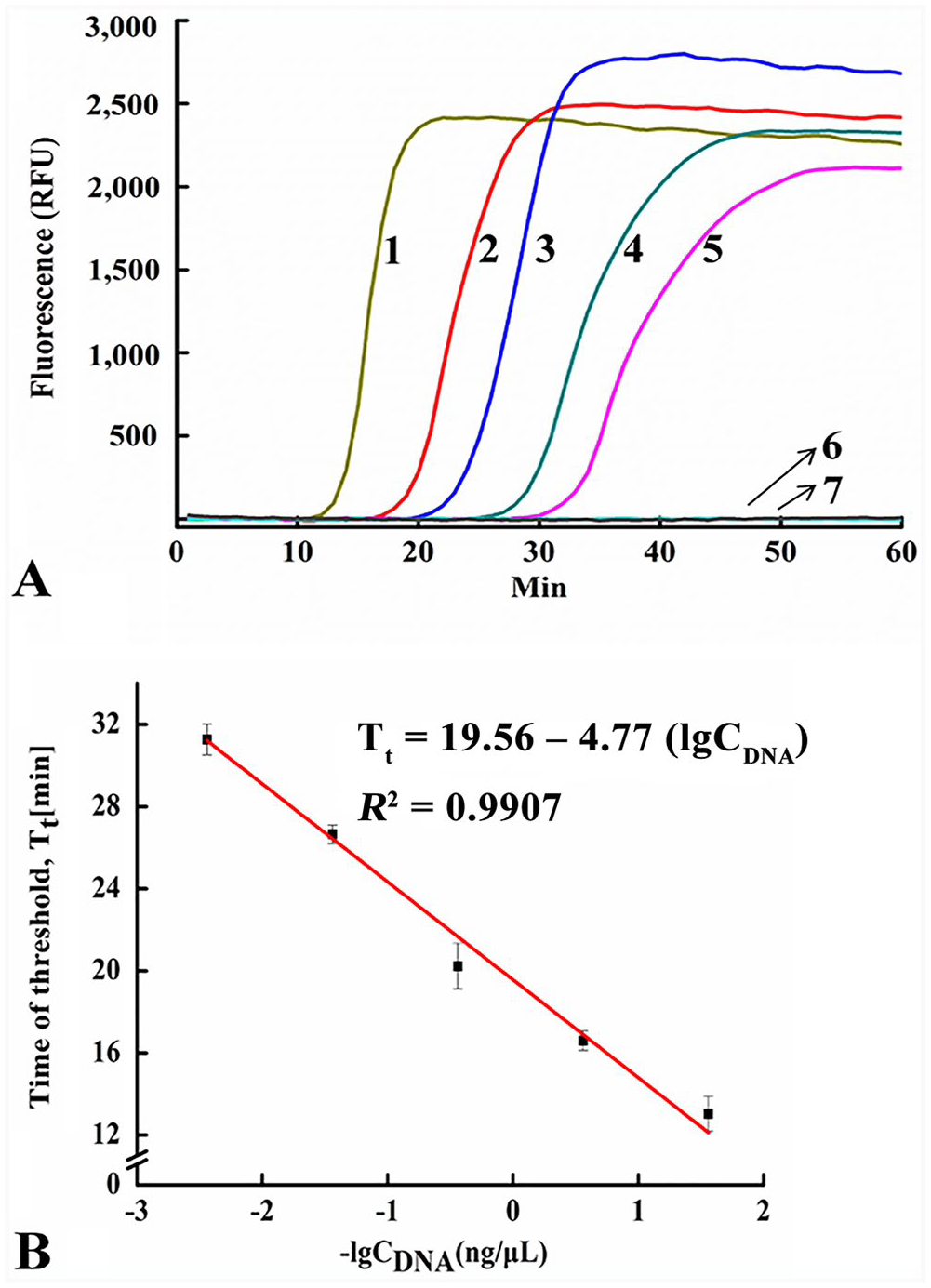
The sensitivity of the SEA method to detect FPLV gDNA.
Specificity of SEA to detect CPV-2 and FPLV
The fluorescence signals of CPV-2 and FPLV began to increase at 19 and 22 min, respectively (Fig. 5), whereas other reactions, including NTC, did not exhibit fluorescence. Additionally, both reactions exhibited the color change after 40 min of amplification reaction (Fig. 5 inset). Nonspecific reactions were not observed, demonstrating the high specificity of the SEA assay, which was particularly important for the differentiation of CPV-2 and FPLV from other pathogens that cause acute enteric disease.
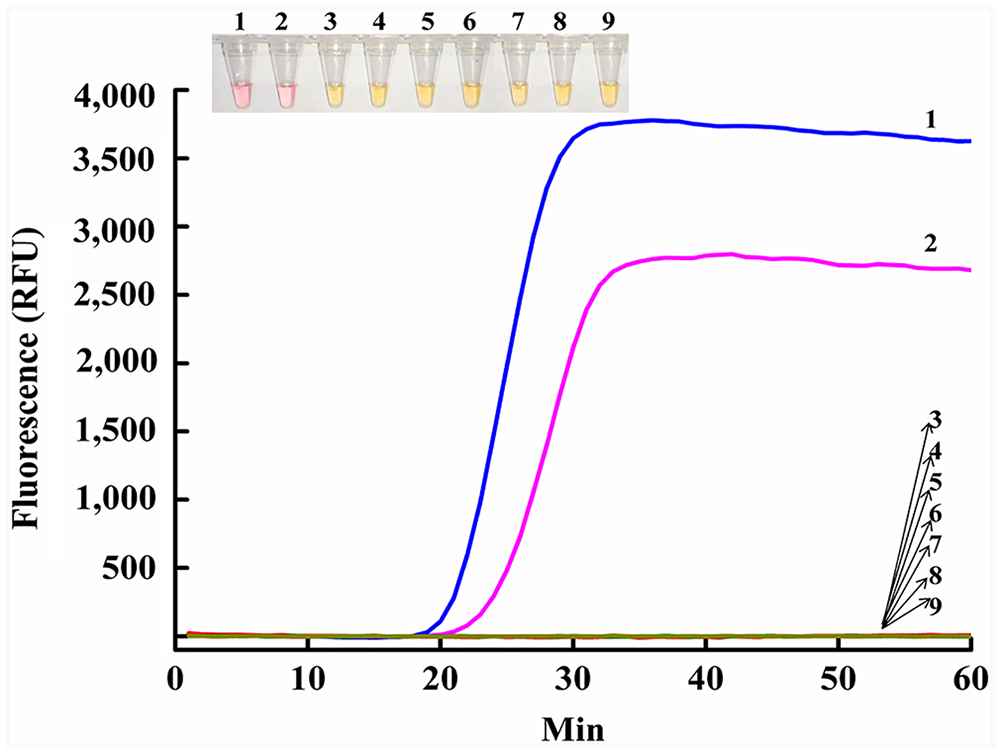
The specificity of the SEA method. In the fluorescence plot, 1 = CPV-2 gDNA; 2 = FPLV gDNA; 3 = F81 cell gDNA; 4 = CCV genomic cDNA; 5 = CDV genomic cDNA; 6 = FCoV genomic cDNA; 7 = FCV genomic cDNA; 8 = gDNA of a negative sample; 9 = no-template control. Inset are the SEA colorimetric reactions, with the same numbering system.
SEA for the detection of different CPV-2 antigenic types and FPLV from fecal specimens
The complete VP2 gene segments of CPV-2 and FPLV were amplified by PCR, and 3 CPV-2 strains were assigned to CPV-2a, CPV-2b, and CPV-2c antigenic types based on amino acid mutations at position 426 (Suppl. Table 3). Except for the NTC, the positive control and 4 clinical samples all had significant fluorescence signals, corresponding 43-bp amplicons appeared in the PAGE gel (Fig. 6 inset, bottom), and there was a visible color change (Fig. 6 inset, top).
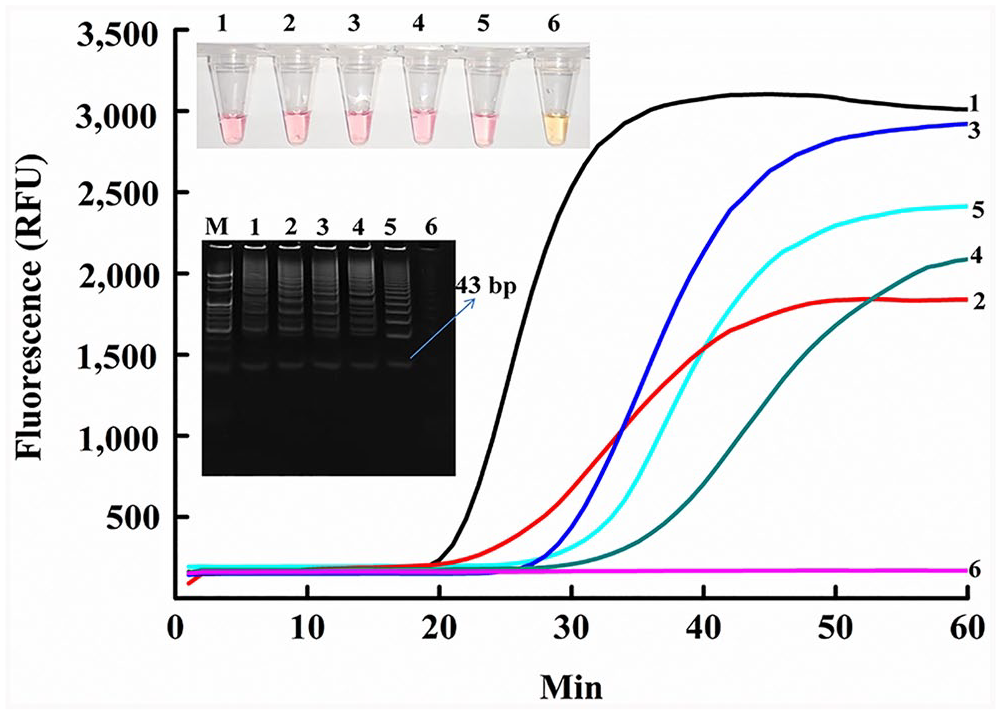
SEA detection of CPV-2 types and FPLV from fecal specimens. All targets were heated for 10 min at 98°C. In the PAGE electrophoresis gel, M = 20-bp marker. In all 6 plots (fluorescence, PAGE electrophoresis, SEA colorimetric method), 1 = CPV-2 positive control; 2 = CPV-2a; 3 = CPV-2c; 4 = CPV-2b; 5 = FPLV; 6 = no-template control.
Comparison of SEA and LAMP
Using the SEA assay, CPV-2 infection was detected in 9 of 35 samples; FPLV was detected in 1 of 18 samples (Table 2). The same results were obtained using the LAMP method. Therefore, both SEA and LAMP exhibited comparable sensitivity and specificity.
Comparison of CPV-2 and FPLV detection by SEA and LAMP directly from fecal specimens.
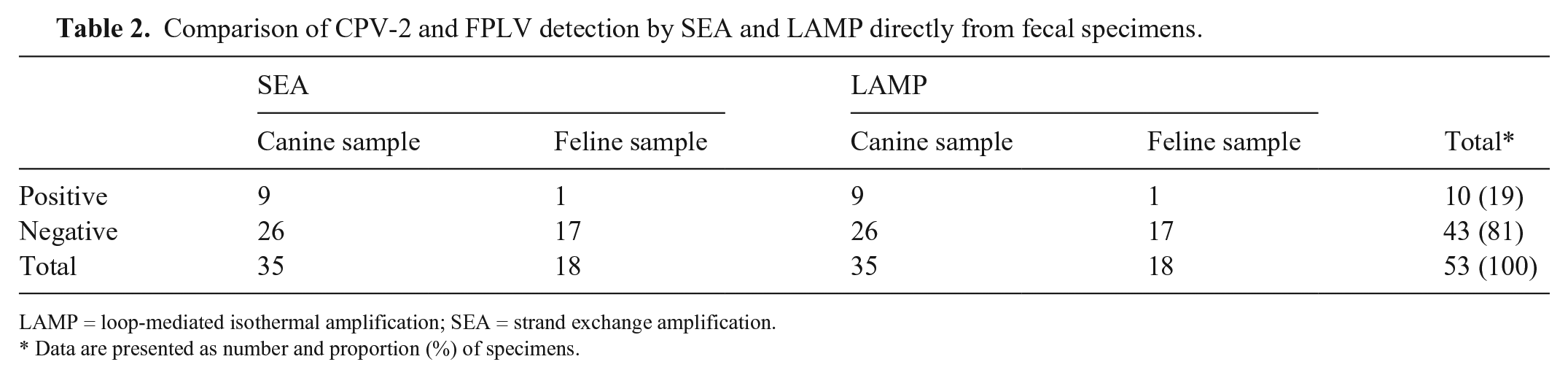
LAMP = loop-mediated isothermal amplification; SEA = strand exchange amplification.
Data are presented as number and proportion (%) of specimens.
Discussion
We used the SEA method to successfully detect the 3 CPV-2 antigenic types (CPV-2a, -2b, and -2c) and FPLV in clinical samples. SEA detected 3.6 pg/μL of CPV-2 gDNA and 6.6 pg/μL of FPLV gDNA, and the results were directly observed by the naked eye. Color reactions have also been used to detect CPV-2 with LAMP, 18 but the reaction tube needs to be opened again to add the chromogenic dye to the reaction system after the completion of the reaction, increasing the possibility of false-positive results because of aerosol contamination. In addition, UV light is required to illuminate the color change. The dye in the SEA colorimetric assay is pre-added, which reduces the chance of aerosol contamination; amplification products are observed directly by the naked eye without extra UV light, which is therefore more cost-effective than LAMP. The success of visual colorimetric detection would contribute greatly to the utility of the SEA method as a tool for point-of-care tests for pets in resource-limited settings.
It has been reported that the rate of nucleotide substitution in CPV-2 (single-stranded DNA) is closer to that of RNA viruses than to that of double-stranded DNA viruses. 20 Therefore, SEA amplification for short regions (40–60 bp) is effective, making it simple to select the most conserved sequence in some viruses with a high mutation rate. LAMP requires a long conserved region to design 6 primers, which may not be possible in the case of some viruses. Fecal shedding of CPV-2 and FPLV has been demonstrated for naturally infected cats and dogs, and fecal sampling is an effective approach for monitoring acute shedding in pets. 7 The SEA method was advantageous given the simple fecal sample treatment and rapid amplification, requiring only a pair of primers and inexpensive equipment.
Supplemental Material
Supplemental_material – Supplemental material for Detection of canine parvovirus and feline panleukopenia virus in fecal samples by strand exchange amplification
Supplemental material, Supplemental_material for Detection of canine parvovirus and feline panleukopenia virus in fecal samples by strand exchange amplification by Mengmeng Liu, Mengzhe Li, Cuiping Ma and Chao Shi in Journal of Veterinary Diagnostic Investigation
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was financially supported by the National Key Research and Development Programs of China (2018YFE0113300) and the National Natural Science Foundation of China (21675094, 31670868).
Supplementary material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.