
Abstract
This consensus document presents the suggested guidelines developed by the Laboratory Technology Committee (LTC) of the American Association of Veterinary Laboratory Diagnosticians (AAVLD) for development, validation, and modification (methods comparability) of real-time PCR (rtPCR) assays. These suggested guidelines are presented with reference to the World Organisation for Animal Health (OIE) guidelines for validation of nucleic acid detection assays used in veterinary diagnostic laboratories. Additionally, our proposed practices are compared to the guidelines from the Foods Program Regulatory Subdivision of the U.S. Food and Drug Administration (FDA) and from the American Society for Veterinary Clinical Pathology (ASVCP). The LTC suggestions are closely aligned with those from the OIE and comply with version 2021-01 of the AAVLD Requirements for an Accredited Veterinary Medical Diagnostic Laboratory, although some LTC recommendations are more stringent and extend beyond the AAVLD requirements. LTC suggested guidelines are substantially different than the guidelines recently published by the U.S. FDA for validation and modification of regulated tests used for detection of pathogens in pet food and animal-derived products, such as dairy. Veterinary diagnostic laboratories that perform assays from the FDA Bacteriological Analytical Method (BAM) manual must be aware of the different standard.
Introduction
Real-time PCR (rtPCR) assays have become the workhorse molecular assays in veterinary diagnostic laboratories (VDLs) since ~2000. PCR assays targeting commonly encountered veterinary pathogens are commercially available (standard test methods) but often laboratories choose to develop their own nonstandard test method (laboratory-developed tests, LDTs). Nonstandard rtPCR assays targeted for use in the laboratory may be completely novel, conversions of conventional PCR assays to a rtPCR format, or extensions of a standard test method outside its intended scope (e.g., new specimen matrix). Whatever the scenario, all nonstandard test methods must be validated for use prior to implementation in an accredited laboratory. In addition, a standard test method, or a previously validated nonstandard test method, may need to be modified in order to accommodate sequence changes within the pathogen, incorporation of a new specimen type or species, or to take advantage of new reagents or instrumentation. Regardless of whether the assay is new or modified, it must be demonstrated to be fit-for-purpose through a predefined process.
Consensus guidelines exist for laboratory assays utilized in human medicine and are regulated by the FDA, which oversees commercial assays18,45 and has proposed guidelines for LDTs. 18 Validation pathways for human medicine exist for both performance 8 and for publication, such as the MIQE and STARD lists.6,7,11,13 Detection of pathogens in veterinary clinical samples is less regulated by federal governments, with oversight provided for some commercial kits through the Center for Veterinary Biologics, U.S. Department of Agriculture (CVB-USDA), but not for LDTs. Thus, it is incumbent upon professional organizations within the veterinary diagnostic community to self-regulate the validation of nonstandard test methods, including LDTs, as well as modifications of validated tests. We provide here an overview of guidance that will help to ensure the quality of rtPCR assays performed in VDLs.
The World Organisation for Animal Health (OIE) has enlisted a consortium of international experts that author the Manual of Diagnostic Tests and Vaccines for Terrestrial Animals, 52 which is a guidance document for VDLs. Several chapters in the OIE manual describe the critical elements of de novo assay development and validation,53–56 as well as critical elements necessary for modification of a validated assay. 57 The American Association of Veterinary Laboratory Diagnosticians (AAVLD) is an organization of 50 U.S. states and 9 Canadian provinces whose diagnostic laboratories are dedicated to providing quality laboratory services for food and companion animals. AAVLD provides an accreditation standard that aligns with ISO/IEC 17025:2017 22 and uses validation guidelines provided in the OIE manual. 53 In addition to AAVLD, ISO 17025, and OIE, 2 other organizations produce standards or guidelines that potentially impact assay validation within a veterinary laboratory. These include guidelines from the American Society for Veterinary Clinical Pathology (ASVCP)16,17,43 and the FDA branch that governs U.S. food safety and oversees the actions of the U.S. Food Emergency Response Network (FERN) and the Veterinary Laboratory Investigation and Response Network (Vet-LIRN). 46
However, there exists a need for more specific guidance covering assay development, modification, and validation of rtPCR test methods. Available guidelines are often considered too expensive to follow,8,11,13 not sufficiently specific (OIE),53–57 or are not relevant for most of the molecular work performed in AAVLD-accredited VDLs.16,17,43,46 The AAVLD Laboratory Technology Committee (LTC) was convened to generate a set of suggested guidelines intended to consolidate the guidance from OIE and to facilitate alignment with the AAVLD Requirements for an Accredited Veterinary Medical Diagnostic Laboratory (https://www.aavld.org/accreditation-requirements-page). To this end, the LTC recommendations are described and compared to OIE guidance and the AAVLD accreditation requirements. The LTC provides herein a set of guidelines that allow validation of a de novo assay and assessment of minor changes in previously validated assays, with the goal of providing a cost-effective means of evaluating the impact of minor modifications. Also described are the requirements that the FDA food safety branch additionally imposes on VDLs.
De novo development of rtPCR
Fit for purpose
The OIE manual provides guidance for nucleic acid assay development and subsequent validation in 5 pertinent chapters.53–57 Here our goal is to review and mesh the guidance from these chapters into one document and offer practical suggestions from the LTC. The scope and intended use of an assay should be determined prior to the start of the assay design process. At the end of the validation process, the developer must assure that the studies support a statement of validity detailing fitness for the intended purpose.
Terms and definitions
To facilitate the discussion of assay development, a glossary of terms is provided (Table 1) comparing definitions provided by OIE 52 and AAVLD. 2 Real-time PCR assay development utilizes the performance criteria of analytical sensitivity and specificity to initially assess the efficacy of the candidate assay on a smaller scale, prior to devoting resources to a full validation. Analytical sensitivity is referred to as limit of detection (LOD) and is obtained by “titrating a known strong positive sample to extinction.” 53 LOD determined by serial dilutions and plotted with log-transformed concentrations gives an approximate linear relationship and yields additional characteristics of dynamic range, linearity, and slope. The slope, in turn, provides PCR amplification efficiency (AE) using the formula: % Eff = 100(10(-1/slope) – 1).8,54 Analytical specificity allows the developer to test susceptibility to specimen matrix inhibitors (selectivity), ability to detect all relevant strains (inclusivity), and the ability to distinguish targeted organisms from genetic near-neighbors and specimen-specific microflora.
Glossary of terms and definitions used in assay validation and verification.
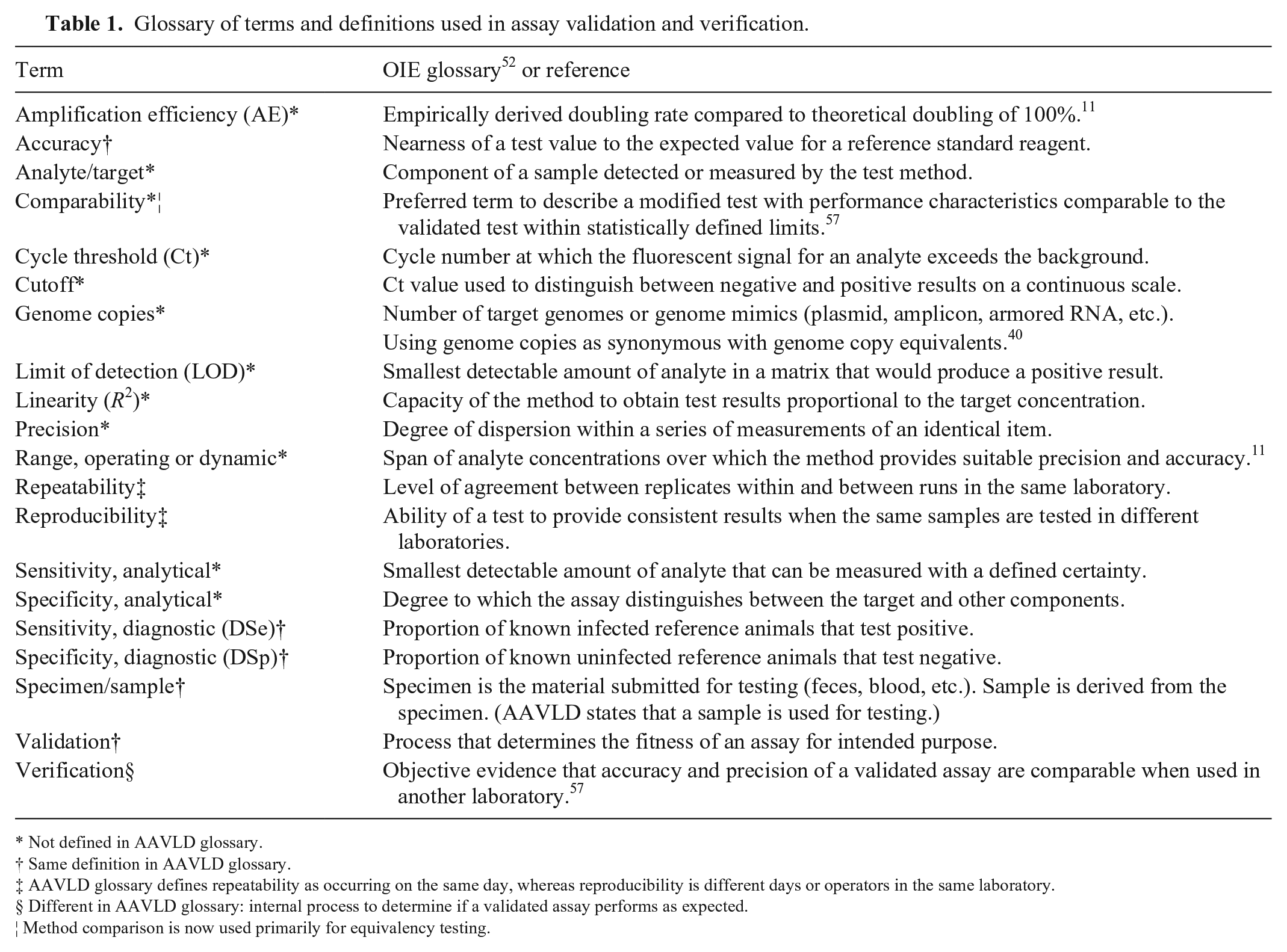
Not defined in AAVLD glossary.
Same definition in AAVLD glossary.
AAVLD glossary defines repeatability as occurring on the same day, whereas reproducibility is different days or operators in the same laboratory.
Different in AAVLD glossary: internal process to determine if a validated assay performs as expected.
Method comparison is now used primarily for equivalency testing.
Development
The assay developer must understand the biology of the pathogen(s) in the host(s) and must match the pathogen target sequence characteristics to the intended use of the assay. Target sequence identification and primer/probe design are the most important steps in developing a useful assay, whether it is conversion of a conventional PCR or development of a new assay. The developer must determine whether the assay will target single or multiple pathogens. Multi-target panels require the use of identical annealing and extension times and usually the same components within a master mix; aside from primer concentration, there is little room for optimization. Once the target gene is selected, additional in silico evaluation of the primer and probe sequences is performed using commercial or publicly accessible software.15,41,44 It is critical to perform a search of nucleotide databanks (https://blast.ncbi.nlm.nih.gov) to determine if target sequences have homology to other agents in addition to the expected target. Design and selection of multiple primer/probe pairs for each target are advised so that the best choice can be made following empirical testing. Basic design guidance10,15,20,31,33,39,42,54 and, more recently, refined primer/probe selection rules are additionally available. 12
Feasibility
In the feasibility study, a limited panel of 6–8 carefully selected samples are tested53,54 in order to limit costs while evaluating which of the candidate primers and probes perform the best. Here the LTC recommendations depart slightly from OIE guidelines, suggesting that the developer initially use a reference strain in buffer as opposed to samples in matrix, until basic efficacy of the primers and probes are established. Determination of the LOD and the operating range requires diluting a very concentrated target suspension through 5–9 ten-fold dilutions in triplicate, which could rapidly deplete a positive sample. The primer/probe set with the highest efficiency, desired LOD, linearity evaluation of R2 close to 1, and spanning the desired range is selected for further evaluation. Then the dose-response curve is repeated in the most likely matrix rather than in buffer to assure that inhibition (selectivity) is considered, similar to OIE guidance. 53 If a partial construct of a gene was used as the target, it is advisable to test native target at this point to ensure that nucleic acid secondary structure does not impede primer and probe binding.12,31 Additionally, performing a melt curve analysis using an intercalating dye such as SYBR green on the selected primers is important to rule out primer–dimer formation empirically.10,20,31
As a next step, the LTC recommends testing 3 or 4 samples of major genetic strains (weak and moderate positives) and 3 or 4 samples containing the most likely commensal competing strains or genetic near neighbors to determine preliminary inclusivity and exclusivity, respectively. It is additionally highly recommended to view PCR as a system and to identify an internal control and extraction method at the outset of the feasibility process.10,12,19,24,58 Concurrently, a positive amplification control, optimally using a traceable reference strain,2,56 should be identified, and sufficient aliquots of this and the negative (no template) control generated to complete feasibility and validation studies.53,54,56 Because rtPCR is either quantitative or semiquantitative, it may benefit a laboratory to calibrate their assay using commercial quantified strains. The OIE suggests use of international or national reference standards to calibrate an assay. 56 The ATCC provides certified reference strains and nucleic acid standards (https://www.atcc.org). The ATCC site also provides links to commercial firms as well as to other national and international sources of standards. Additional recommended controls for routine performance of PCR are discussed in accompanying articles found in this focus issue.44,58
De novo validation
Once an assay is developed, validation commences and is carried through 4 stages of the validation pathway as outlined in the OIE guidelines. 53 In stage 1, analytical sensitivity, specificity, and preliminary repeatability are determined. Stage 2 involves evaluation of the assay using diagnostic samples. Stage 3 is comprised of more extensive repeatability and reproducibility evaluation. Stage 4 involves establishing practical considerations for implementation of the assay.
Stage 1, analytical characteristics
Several methods can be used to determine the LOD, including 10-fold dilution series, 2-fold dilution series,53,57 or probit analysis.8,24,55 The simplest and most economical method to determine LOD for a rtPCR assay is by utilizing the dilution series that was generated during the analytical sensitivity determination. The final 10-fold dilution for which all 3 replicates are positive provides a conservative LOD estimate provided it was determined in the sample matrix intended for use. Using a hypothetical example, 3 of 3 independent 10-fold dilution series consisting of known target genome copies could detect 10 copies of the target, but only 1 of 3 of the dilution series could detect the target at 1 genome copy. With this example, the LOD would be 10 genome copies.
In general, the number of replicates for LOD determination of a de novo assay validation ranges from 3 to 12 replicates, with the higher number required in some statistical analyses. 8 The replicates may be 10-fold dilutions or, with a greater expenditure of resources, 2-fold dilutions. An example of a LOD determined by a 2-fold dilution series is provided by the developers of a Mycoplasma cynos assay described in this focus issue (Table 2). 40 Twelve replicates for each of 8 two-fold dilutions gave an LOD of 10 genome copies (i.e., the last dilution in which all replicates were positive; Table 2). These same data can be used for probit or logit analysis by applying generalized linear models to the results of the dilution series experiment. 8 Based on the concentration-response curve generated (Fig. 1), the concentration in which 95% of targets are positive is 4.6 genome copies (anti-log2 of 2.2). It is important to note that, in this example, the concentration corresponding to the estimated LOD from the probit analysis was not tested directly.
Determination of LOD based on 2-fold dilution series for Mycoplasma cynos assay. 40
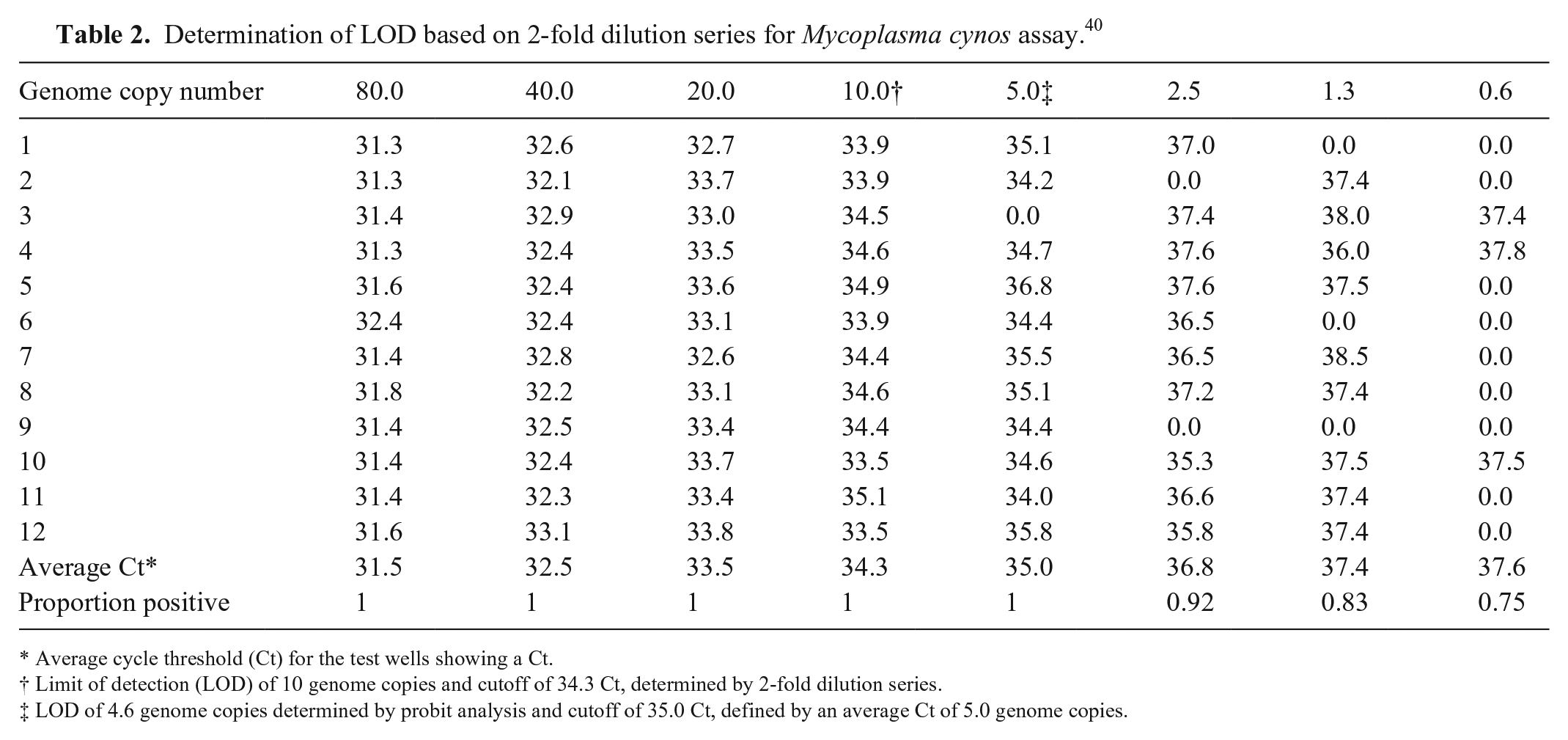
Average cycle threshold (Ct) for the test wells showing a Ct.
Limit of detection (LOD) of 10 genome copies and cutoff of 34.3 Ct, determined by 2-fold dilution series.
LOD of 4.6 genome copies determined by probit analysis and cutoff of 35.0 Ct, defined by an average Ct of 5.0 genome copies.
Cutoff
The cutoff value provides classification of results as either positive or negative and may also include an indeterminate range. The target nucleic acid dilution series detailed above generates a standard curve that allows for accurate quantification of target genome copies. In VDLs, generally a single positive amplification control (PAC) concentration is routinely included for a rtPCR assay, thus limiting the precision of quantification, and making most of the assays used semiquantitative rather than absolute. Thus, the mean cycle threshold (Ct) value of the lowest concentration reliably detected in the dilution series may become the cutoff metric used to interpret diagnostic assay results on a daily basis. For a LOD determined by probit analysis, the cutoff is defined as the mean Ct of the next most concentrated dilution tested to the LOD defined by probit. In the example in Figure 1, the average Ct value is 35.0 at a genome copy of 5, which is the tested value at the concentration immediately above the probit value.
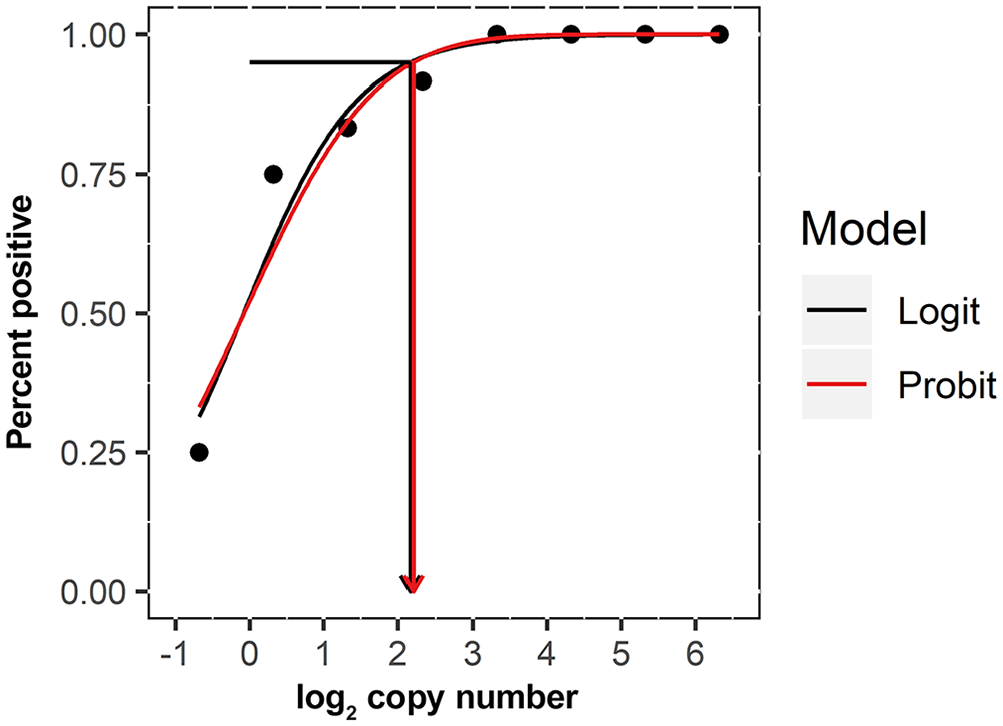
Probit and logit analysis (R Core Team) based on the concentration-response curve generated from the proportion positive results (Table 2). The concentration at which 95% of targets are positive for logit is 4.5 genome copies (95% CI: 3.0, 6.8) and for probit is 4.6 genome copies (anti-log2 of 2.2; 95% CI: 3.1, 6.9).
Analytical specificity
In the feasibility stage, a preliminary assessment of specificity was performed to aid in vetting the primer selection. Once validation commences, a more thorough evaluation of inclusivity, selectivity, and exclusivity is warranted. Traceable isolates of major strains are used for LOD determinations to assess inclusivity and to be compliant with the AAVLD accreditation requirements. Multiple matrices are evaluated to determine selectivity, and traceable genetic near neighbors of strains of commensals are tested to evaluate exclusivity. Examples of studies with thorough analytical specificity studies are referenced.34,40,50
Repeatability
Repeatability is a measure of agreement among replicates of a sample within runs and between runs in the same laboratory. There are many sources of variation that will affect the outcome or result of an assay within a laboratory, variation among operators being one of these factors. The guidance provided by the LTC recommends that one operator perform this comparison, which differs slightly from the OIE, which recommends that testing should be done by more than one operator. 53 To extend the LTC recommendation, additional operators within a laboratory perform repeatability in order to show competence during their training and, through this practice, assay repeatability measures become congruent with the OIE guidelines. The LTC recommends testing 3 concentrations (high, medium, and low), which is consistent with other guidelines including OIE, 53 and to test 5 replicates of these concentrations in 6 separate runs. 32
The LTC studies recommended to obtain stage 1 analytical sensitivity, analytical specificity, and repeatability data with acceptance criteria are summarized and compared to OIE guidelines (Table 3). In general, recommendations are similar, although some suggestions from the LTC provide additional guidance. For example, LOD determination involves testing across the operating range of the assay, usually requiring 5–9 log10 dilutions. 8 Detailed examples of analytical and diagnostic validation are referenced.40,50,51
Recommended studies for validation of a newly developed real-time PCR (Stage 1, analytical characteristics; Stage 2, diagnostic characteristics; Stage 3, reproducibility).
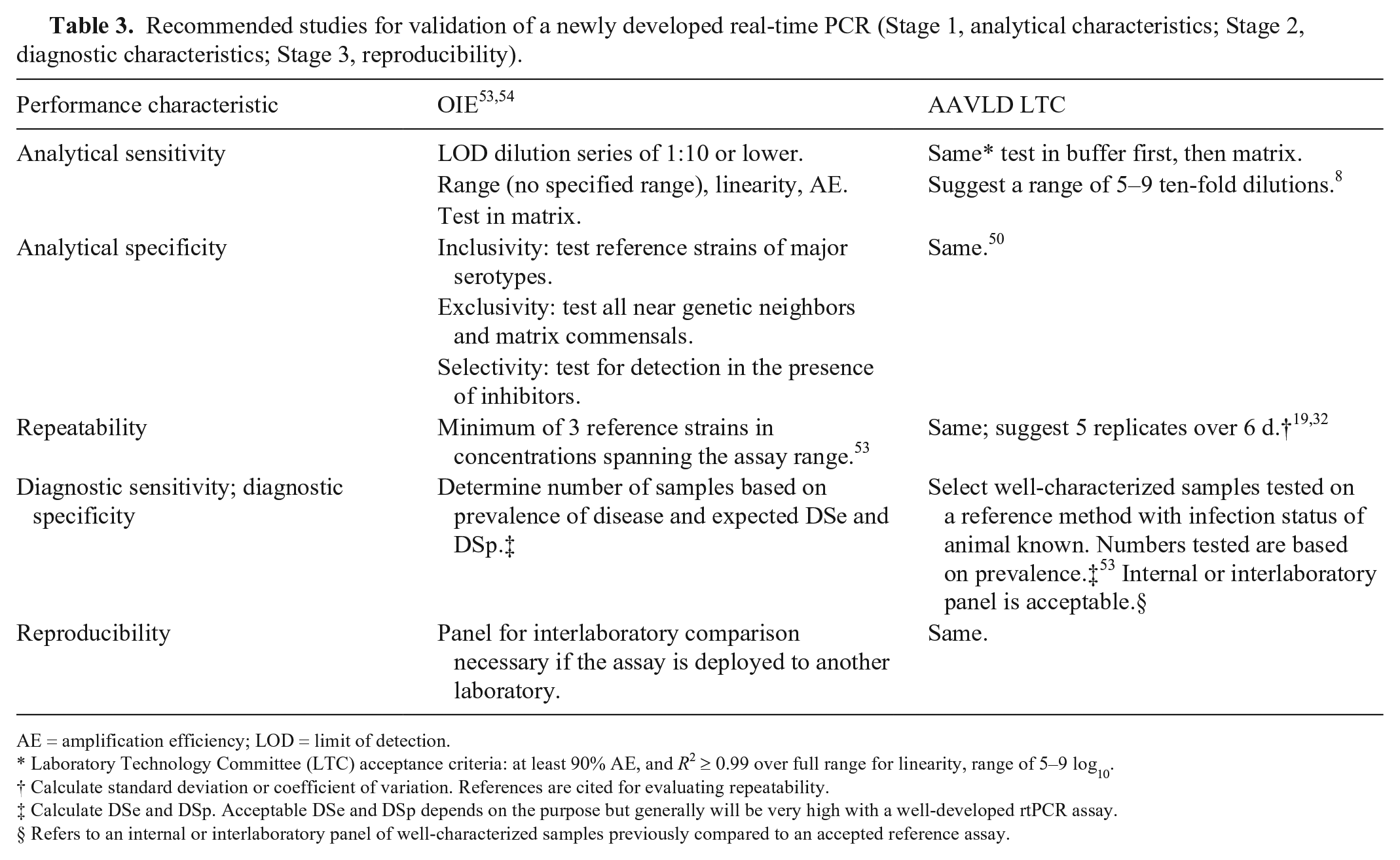
AE = amplification efficiency; LOD = limit of detection.
Laboratory Technology Committee (LTC) acceptance criteria: at least 90% AE, and R2 ≥ 0.99 over full range for linearity, range of 5–9 log10.
Calculate standard deviation or coefficient of variation. References are cited for evaluating repeatability.
Calculate DSe and DSp. Acceptable DSe and DSp depends on the purpose but generally will be very high with a well-developed rtPCR assay.
Refers to an internal or interlaboratory panel of well-characterized samples previously compared to an accepted reference assay.
Stage 2, diagnostic characteristics
In stage 2, diagnostic sensitivity (DSe) and diagnostic specificity (DSp) are determined. Recommendations for the number of samples needed for a full validation are found in the OIE guidance. 53 The analytical cutoff should be established from the LOD (determined by 2-fold, 10-fold, or probit/logit methods) then applied to a reference diagnostic sample set in order to verify the cutoff Ct value.
The source and quality of samples used in the validation process is a key element to reliably evaluating diagnostic performance characteristics. The acquisition of samples should be described and fully documented to include source, storage, collection, transfer, transport media, preservative used for storage, and relevant animal information (breed, age, sex, disease status, clinical history). 53 Random selection of samples will guarantee good representation and help avoid bias; 25 however, it is critical to include representatives of all host species and all sample matrices.53,55 Controversy exists as to whether any of the samples used during the diagnostic performance evaluation can be spiked samples (inoculated with known quantities of a pathogen). This is discussed for assays pertinent to wildlife species in a companion article in this focus issue. 23
In the absence of knowledge about the true disease status, statistical methods can be used to estimate DSe and DSp.21,30,47 Validating assays for wildlife often require alternative statistical methods, as reviewed in this focus issue. 23
The LTC recommends studies to obtain stage 2 DSe, DSp, and repeatability data with acceptance criteria, as summarized and compared to OIE recommendations in Table 3. Note that in performance of the stage 2 studies, operators should be blinded to the true status of samples. Examples of studies performed using the LTC stage 2 validation guidelines are referenced.40,51
Stage 3, reproducibility
Stage 3 is characterized by establishment of reproducibility, which is the assay characteristic that measures consistency of test results derived from assays performed in different laboratories using aliquots of the same samples. 53 This is a critical step for LDTs used outside of the originating laboratory. In order to establish reproducibility, a panel of well-characterized samples representing a range of concentrations and all possible matrices is collected and tested using the reference method. These samples should be aliquoted and stored to maintain stability and homogeneity. Assays developed for internal use may be tested using a reference panel and in an alternative laboratory in order to fully align with the OIE pathway. The designated reference panel can also serve as a reference panel for determining accuracy following any subsequent assay modifications. One-time comparison with a reference panel, documentation of “internal or interlaboratory comparison to an accepted methodology and protocol,” together with “ongoing documentation of internal or interlaboratory performance using known reference standards for species and/or diagnostic samples of interest” is one way to fulfill test method validation per version 2021-01 of the AAVLD accreditation requirements. 2 The LTC recommendations for reproducibility are compared to OIE guidance in Table 3.
Stage 4, implementation
Implementation includes the practical steps of bringing an assay on board: cost accounting, management approval, finalizing the standard operating procedure (SOP), control chart establishment, incorporation into laboratory information management systems, etc. Interpretation criteria should be established prior to launching the assay. Test results must be considered in context of “fitness for purpose.” Factors to consider beyond the laboratory aspects of test validation include: presence or absence of clinical disease; whether the agent is transboundary, rare, or endemic; possibility of environmental contamination; and the potential of detecting vaccine components. For example, the rtPCR Ct value may be interpreted differently if the animal was known to be recently vaccinated with live 49 or inactivated vaccines. 38
Once the test is launched, assays must be continually monitored by the use of daily in-house reference controls. All guidelines concur on the necessity of having a positive amplification control and a “no template” amplification control for each target; internal controls are strongly recommended. All controls are reviewed in companion articles in this focus issue.44,58 Establishment of criteria to monitor assay performance is needed to complete the OIE validation pathway and to meet the AAVLD accreditation requirement. 2 In addition to daily monitoring of the assay, primer and probe sequences should be monitored routinely to determine whether emerging/novel nucleotide sequence mutations in the target region alter the performance of the assay. 15 As a final note, tests offered to a client must be completely validated, and the test method should be communicated with the client on the test report. If the client requests use of a non-validated test method (e.g., use on a new species or specimen matrix) and the laboratory conducts such testing, there must be a disclaimer statement added to the test report in order to be compliant with AAVLD accreditation requirements.
Method comparability
A validated assay in routine use may need to be modified. In keeping with the OIE naming convention, 57 the LTC refers to this as method comparability rather than comparison. Modifications include reagent replacement, platform changes, or extraction modifications.32,57 The LTC considers changes to the primer or probe sequence to be major changes, although OIE guidelines suggests that changes within the same amplified region are minor. The OIE guidance considers species or matrix additions to be major modifications to an assay. Box 1 shows the consensus between OIE and LTC designations of minor modifications. Under optimal circumstances, a direct comparison between the reference procedure and the modified procedure, hereafter referred to as the candidate method, is desirable. The procedures described are intended to make a direct comparison, but without the stringency of demonstrating statistical equivalence, which would require defining margins that are related to irrelevant scientific differences.3,37 The recommended testing should reveal any egregious changes to the performance characteristics of the modified assay. The LTC recommended method comparability studies for acceptance of a modified rtPCR assay are compared to OIE guidance in Table 4.
Minor modifications of a PCR assay.

Recommended method comparability studies for acceptance of a modified real-time PCR assay.
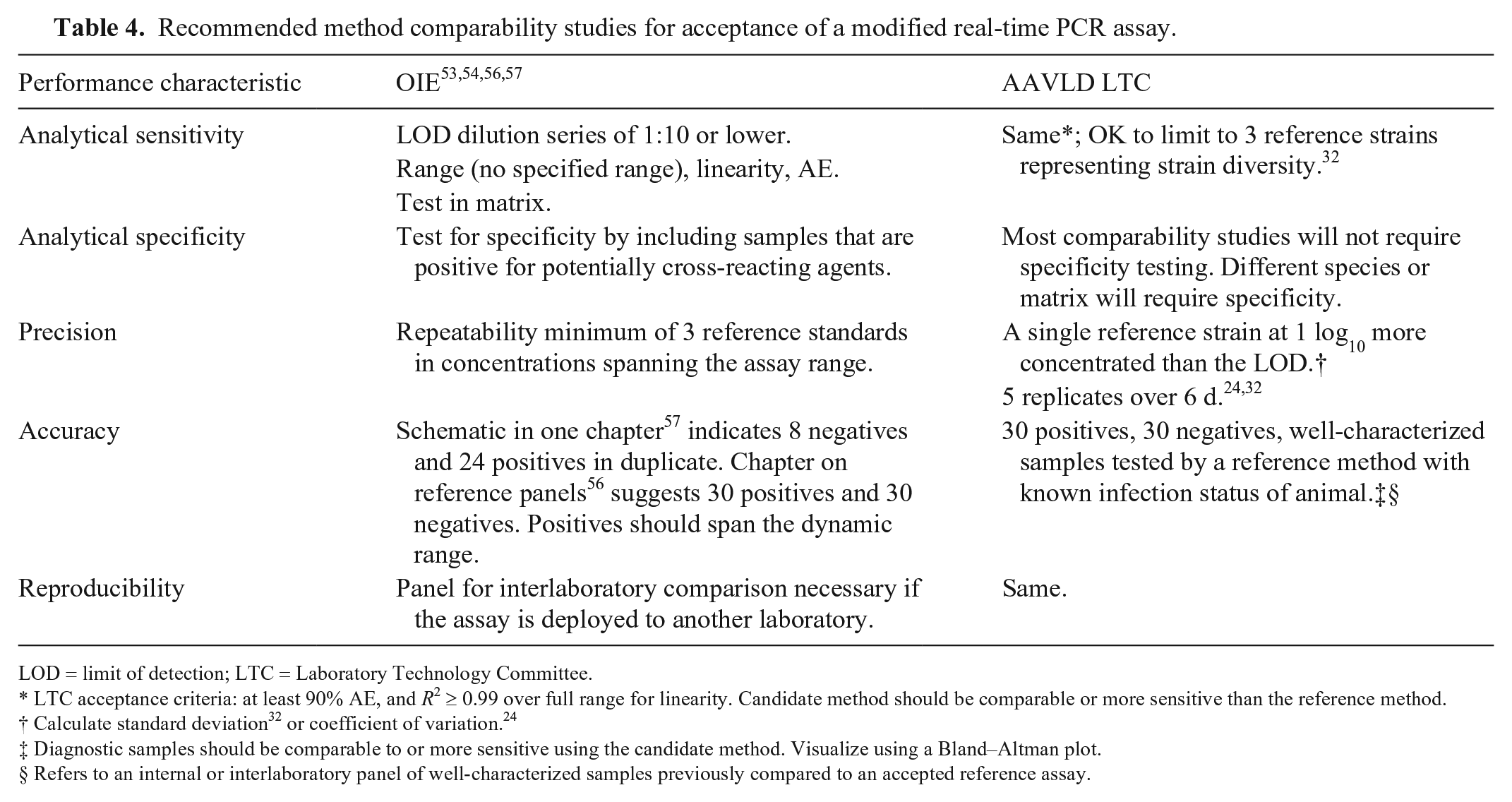
LOD = limit of detection; LTC = Laboratory Technology Committee.
LTC acceptance criteria: at least 90% AE, and R2 ≥ 0.99 over full range for linearity. Candidate method should be comparable or more sensitive than the reference method.
Diagnostic samples should be comparable to or more sensitive using the candidate method. Visualize using a Bland–Altman plot.
Refers to an internal or interlaboratory panel of well-characterized samples previously compared to an accepted reference assay.
Method comparability analytical characteristics
An analytical sensitivity study is recommended using up to 3 reference strains, 32 which is a reduction in resources from the requirement to test all major strains in a de novo assay. The 3 reference strains selected should span the sequence diversity. 32 An analytical specificity study is not necessary but can be performed optionally unless a change of the sample matrix or species is evaluated, which would necessitate specificity testing.
The repeatability of the assay is assessed by conducting a study to estimate the intra- and inter-assay variance using one reference strain. Testing 3 concentrations, as recommended for a de novo assay, is one of the most resource-intensive steps in the validation process, with little value realized from testing strong and mid-range concentrations. Thus, our recommendation is to test a low concentration that is within the linear range of the assay but near the LOD. The test material is prepared in advance in bulk such that 1 aliquot is tested 5 times on each of 6 independent testing occasions32,48 by 1 operator. To analyze the findings statistically, a linear mixed-effect model 29 can be used to estimate the inter-assay variance (between testing occasion) and the intra-assay variance (within testing occasion variance).9,48 To analyze the results with more readily accessible statistics, standard deviation or coefficient of variation can be used. Examples of acceptable standard deviation and coefficient of variation are referenced in Table 4.
Method comparability accuracy
Sample size determination is not stated for comparability; however, a schematic suggests 8 negatives and 24 positives in duplicate is sufficient; 57 a panel of 30 positive and 30 negative samples for assay evaluation is recommended by the LTC and is referenced in the OIE guidance. 56 Ideally, these samples are field diagnostic samples that are tested using the reference assay method (gold standard) because these can then become the reference panel needed to be congruent with the OIE guidance. The intention is to test enough samples to span the operating range of the assay. The operator should be blinded to sample status. In a side-by-side comparison, aliquots from the same sample should be tested concurrently (in the same test session) using the reference and candidate method. To assess the findings, the data can be plotted using a square correlation plot27,28 with modification 32 or a Bland–Altman plot.4,5,26 The paired mean difference can be estimated when appropriate. For rtPCR-based assays, this would be reduced to the number of samples in which a Ct was observed using both methods. The correlation and concordance correlation coefficients along with confidence intervals can also be estimated using the samples that produce a response for both methods. 28 Finally, kappa can be estimated, and a confidence interval provided for the assay comparison. 1
A primary impetus for formation of the LTC was to generate guidelines that limit the expenditure of resources needed to re-validate an assay. To this end, side-by-side testing may be unnecessary because of either the nature of the modification or use of a reference panel. For example, cross-contamination analysis is necessary when robotic equipment is evaluated to replace manual procedures but would be difficult to set up in a parallel situation performed manually. In other cases, a well-characterized panel of samples already tested by the reference method could be tested with the candidate method without repeating the reference testing concurrently, as long as care is taken to preserve the integrity of the original sample. Values near the cutoff may be lost in the reference sample due to repeated freeze–thaw cycles, so it is important that aliquots of the reference panel undergo the same freeze–thaw and storage conditions. 56 Any discrepancy between the candidate and reference assay would require repeating all samples within a similar quantitative range side-by-side. This reference panel may be a set of internal well-characterized samples, or a panel generated by interlaboratory collaboration, both of which are an option for validation according to the AAVLD standard. 2 It is expected that the candidate assay yields the same or better Ct value than the reference assay for the panel samples.
Given the limited number of samples used, a method comparability experiment is not intended to estimate the DSe and DSp of an assay. As such, the findings should be summarized. Should the modified assay result in a more sensitive assay, the analyst could sequence the amplicon to resolve whether the additional detections are specific to the target, as discussed in a companion article in this focus issue and elsewhere.14,15
Verification
Most guidelines, whether ASCVP, OIE, or FDA, have essentially the same definition of verification; however, different amounts of testing are required to complete a verification study. Verification studies typically demonstrate that an assay performs as expected when newly adopted without changes in a laboratory. Implementation of a commercial kit or of a published assay are examples of verification. Verification studies are sufficient provided the assay was fully validated by the company or the authors of the published article. The original validation data for a kit should be available from the company; published articles should have followed one of several accepted guidelines for validation studies.6–8,11,53 To perform a verification, analytical sensitivity (LOD) is recommended by the LTC and performed by testing 10-fold dilutions of a single reference strain in matrix; no further analytical sensitivity or specificity testing is suggested. Accuracy for verification as interpreted by the LTC involves testing a minimal number of well-characterized diagnostic samples, specifically 30 positives and 30 negatives previously assessed by a reference method. 56 Although a verification study is appropriate to introduce a USDA-regulated commercial kit into use in a laboratory, it should be noted that these guidelines are not intended to be used to validate changes to commercial kits that are licensed by the CVB-USDA. The kit must be used as approved by the CVB.
Note that verification as defined by AAVLD accreditation requirements includes studies performed to obtain objective evidence to show that a changed standard method can be performed properly. The LTC recommendations concur that verification is needed for a changed standard method that comes from a governing body. However, if the modification is initiated within the laboratory, then additional work, more in line with method comparability, is recommended.
Special cases of comparability
Addition of an internal control
It is highly recommended that rtPCR assays incorporate either an exogeneous or endogenous internal control. That said, many laboratories do not currently have these incorporated in their assays for economic reasons. Use of an internal control is a special case of comparability testing because the assay is modified from a singleplex to a duplex assay. An example of incorporation of an internal control is given in a companion article. 58
Conversion of singleplex to multiplex
Real-time PCR assays with multiple targets provide cost savings and can be performed without a loss of sensitivity. The researcher can assess the process (extraction and PCR assay) starting with stocks of pathogens or can assess the assay itself starting with nucleic acid. Here we give an example starting at the extraction step. Careful choice of extraction kits allows co-extraction of DNA and RNA agents. Inclusion of a reverse-transcription step allows co-amplification of DNA and RNA agents as well. Each assay is validated initially as singleplex and then combined in a multiplex. The critical parameter to test is whether a single component assay is still functional across its established range in the presence of concentrated amounts of potentially competing assays. Each singleplex assay is compared with the corresponding assay in the multiplex format and also compared to an assay with competing targets and assessed for performance criteria of dynamic range, linearity, and LOD (Fig. 2). Precision is determined by testing replicates of a single concentration in the multiplex series using the dilution closest to 33–34 Ct for all targets (e.g., ~1 log more concentrated than the LOD). Sufficient volume of the chosen dilution should be prepared to complete all necessary testing, and aliquots should undergo the same storage and freeze–thaw conditions. 32 Each mixture is tested 5 times within each of 6 independent runs and analyzed as described previously. 32
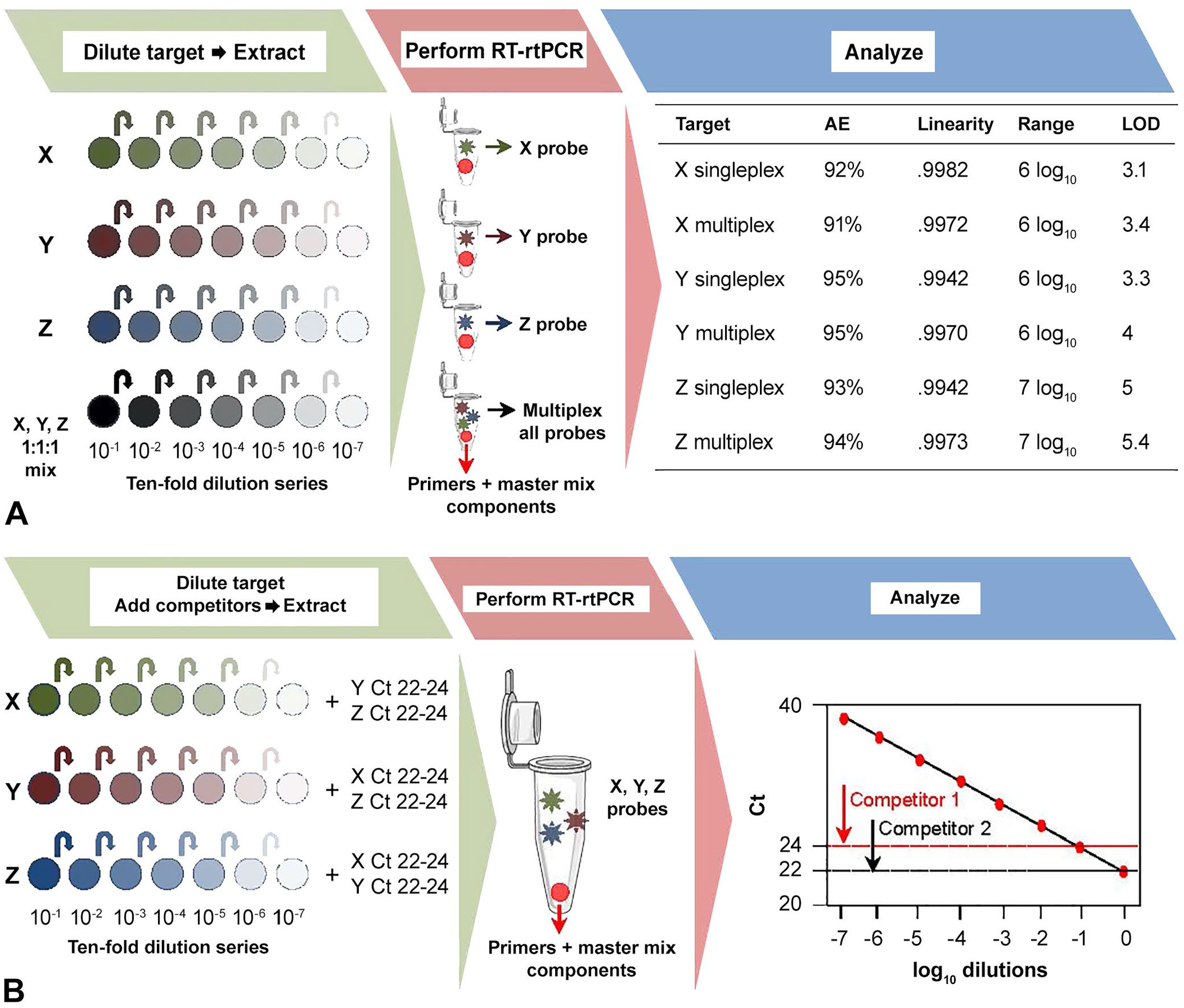
A reference set of well-characterized diagnostic samples, ideally 30 positive samples with various target concentrations and 30 negative samples per analyte, 56 is used to assess the diagnostic performance of the multiplex assay. The number of reference samples can be reduced if specimens from animals with natural polymicrobial or multiple viral infections are tested to verify the multiple-pathogen detection capability of the assay. Also, to be congruent with the AAVLD standard, an internal or interlaboratory panel of well-characterized samples can be used without the necessity of a side-by-side comparison. The recommendation from the OIE guidance is to test a subset panel (5–8 of each species or matrices) when faced with a potentially large sample set. 57 The modified assay is considered acceptable if the results of the subset tested with the candidate method are equal to or better in performance compared to the reference method. Methods for resolution of discordant samples should be identified at the beginning of the study as described earlier. Secondary assays such as sequencing can be used to confirm specificity of positive samples. Cutoffs should be the same for each assay in singleplex or multiplex format. Multiplex assays developed by AAVLD member laboratories using the OIE and LTC guidelines are referenced.34,35
Modification of multiplex assays
Special cases of multiplex re-validation may arise (Table 5). Changes made to a validated multiplex assay, such as PCR chemistry, platform, minimal nucleotide changes, etc., may be assessed as described previously using a methods comparability study. 32 Host species and matrix introductions may require a more extensive re-validation, which can first be assessed using a reference panel of well-characterized samples. 57 Dependent on the modification, the testing may utilize a methods comparability assessment of candidate multiplex to the original multiplex assay. Other types of re-validation include incorporation of a newly developed singleplex into a validated multiplex, incorporation of additional validated singleplex(s), or performance of a partial multiplex.
Multiplex assays modifications requiring method comparability.

Can also utilize an inter- or intra-laboratory reference panel to demonstrate accuracy for the modified multiplex assay.
Method modification guidelines from FDA
Most state VDLs provide testing for foodborne disease pathogens in animal-derived products or feeds and work closely with their public health counterparts in FERN and Vet-LIRN. Both of these organizations utilize methods found in the U.S. FDA’s Bacteriological Analytical Method (BAM) manual. The FDA Food Programs Governance Board regulates the methods used in food safety and is a different branch of government than that which regulates human diagnostic laboratory procedures.
As noted throughout the description of de novo validation, the LTC recommendations closely follow those of the OIE. The U.S. FDA recommendations, however, are different, largely because of the nature of “samples” with a nearly unlimited number of matrices. With food items, spiking in order to simulate a contaminated food is the only option, given that no archive exists that could accurately represent all of the food products subject to spoilage and subsequent outbreak testing needs. Compared to the OIE and LTC guidance, requirements for validation of an assay are more exacting, whereas requirements for modification such as extension to a new matrix are less prescriptive.
The most likely situation in which a veterinary laboratory may need to follow U.S. FDA guidance is when testing a food product associated with a submission from an outbreak, but the food product is not a validated matrix. Recent guidelines 46 outline requirements for validation by a single laboratory during emergency situations (L1), or non-emergency (L2) situations, and validation for use in ≥2 laboratories (L3) or 8–10 laboratories (L4; Box 2). Most VDLs would modify BAM methods for use with a new food matrix using level 1 or 2 criteria. A comparison between the LTC recommendations for assays targeting animal pathogens and FDA requirements for a modification to BAM methods in a single laboratory (L2) is shown (Table 6) for prokaryotic pathogens and with non-microbial targets (FDA equates “microbial” to prokaryotes only). Acceptance of the modification would require at least 50% of the replicates at the LOD, that all of the replicates at the higher concentration were detected, and that no target was detected in the uninoculated samples. Note that adherence to the FDA guidelines are requirements not suggestions. Adhering to both FDA and the AAVLD standard is possible but ultimately will be determined by an individual laboratory’s quality system.
FDA levels of validation.

U.S. FDA requirements for studies to assess performance for modified BAM assays.

BAM = Bacteriological Analytical Method manual.
U.S. FDA: https://www.fda.gov/media/83812/download
U.S. FDA definition of microbial equates to prokaryotic (bacteria); non-microbial would be parasites, fungi, viruses, etc.
American Society for Veterinary Clinical Pathology
The American Society for Veterinary Clinical Pathology (ASVCP) also provides guidance for clinical pathology laboratories that are usually housed within a veterinary school. Disciplines covered include hematology, cytology, clinical chemistry, toxicology, and serology. ASVCP has been regularly active in providing guidelines for their members;16,17,43 however, the disciplines targeted are quite different from the primarily infectious disease testing that a state VDL performs, with very little rtPCR performed in a clinical pathology laboratory. Genetic testing for breed identification or tumor markers is within the purview of clinical pathology; however, few if any laboratories abiding by ASVCP guidelines provide this service. Guidelines have been adopted for canine clinical genetic testing laboratories from human genetic testing and are in a nascent stage, 36 with genetic testing usually performed in private laboratories.
Alignment with AAVLD accreditation requirements
The AAVLD requirements for validation are summarized here. We reiterate the comparison with emphasis on how the LTC recommendations comply or extend beyond the AAVLD accreditation requirements.
First, all test methods validated and in use in a laboratory must be constantly monitored by use of a traceable reference control through ongoing documentation of internal or interlaboratory performance. The LTC recommendations are in accordance with this requirement by requiring a reference PAC for each target assay.
Second, one of the following 3 criteria, paraphrased here, must also be met:
endorsed or published protocol by a reputable technical organization;
published peer-reviewed article with sufficient documentation; or
comparison to an accepted method through internal or interlaboratory comparison.
Implementation of the USDA National Animal Health Laboratory Network (NAHLN) assays are an example of the first bullet point. The LTC recommendation and AAVLD accreditation standard concur in that a reference panel, such as a required proficiency test, would be sufficient for verification prior to implementing a NAHLN assay. The second bullet point, adoption of a published assay, would require verification, which could be accomplished by testing an inter- or intra-laboratory panel of well-characterized samples or by using another process that can objectively demonstrate accuracy of the method in obtaining the expected results. The third bullet point applies to either de novo or method comparability. The LTC recommendations again fit with the AAVLD accreditation requirements as long as testing of an inter- or intra-laboratory panel of well-characterized samples used an accepted methodology and generated objective evidence confirming fitness for purpose.
Footnotes
Acknowledgements
We thank the number of individuals who contributed to the discussions and the development of these guidelines over a number of years. We specifically thank Amy Glaser, Deep Tewari, Beth Harris, Kate Schumann, Susanne Hinkley, Pam Ferro, and Megan Schroeder. We also thank Mauricio Navarro for his expert help with ![]() .
.
Declaration of conflicting interests
The authors declared no potential conflict of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.