
Abstract
Parapoxviruses (PaPVs) cause widespread infections in ruminants worldwide. All PaPVs are zoonotic and may infect humans after direct or indirect contact with infected animals. Herein we report the development and validation of a highly sensitive real-time PCR assay for rapid detection of PaPVs. The new assay (referred to as the RVSS assay) was specific for PaPVs only and had no cross-reactivity against other pox viruses. Using a recombinant plasmid as positive control, the analytical sensitivity of the assay was determined to be 16 genome copies of PaPV per assay. The amplification efficiency estimate (91–99%), the intra- and interassay variability estimate (standard deviation [SD]: 0.28–1.06 and 0.01–0.14, respectively), and the operator variability estimate (SD: 0.78 between laboratories and 0.28 between operators within a laboratory) were within the acceptable range. The diagnostic specificity was assessed on 100 specimens from healthy normal animals and all but 1 tested negative (99%). The diagnostic sensitivity (DSe) was assessed on 77 clinical specimens (skin/scab) from infected sheep, goats, and cattle, and all tested positive (100%). The assay was multiplexed with beta-actin as an internal positive control, and the multiplex assay exhibited the same DSe as the singleplex assay. Further characterization of the PaPV specimens by species-specific real-time PCR and nucleotide sequencing of the PCR products following conventional PCR showed the presence of Orf virus not only in sheep and goats but also in 1 bovid. The validated RVSS assay demonstrated high specificity, sensitivity, reproducibility, and ruggedness, which are critical for laboratory detection of PaPVs.
Introduction
Parapoxviruses (PaPVs; genus Parapoxvirus, family Poxviridae) include 4 species, Bovine papular stomatitis virus (BPSV), Orf virus (ORFV), Parapoxvirus of red deer in New Zealand (PVNZ), and Pseudocowpox virus (PCPV). 5 The most common hosts of PaPVs are ruminants, including sheep and goats (infected by ORFV) and cattle (infected by BPSV and PCPV). In addition, PaPVs have also been detected in wildlife, including seals, red squirrels, and Japanese serows. 5
All PaPVs are known to be zoonotic, infecting humans after direct or indirect contact with infected animals. 1 The Centers for Disease Control and Prevention listed ORFV infections among the zoonotic diseases in humans (http://goo.gl/WyflEj). Human PaPV infections can resemble localized infections with orthopoxviruses (OPVs), such as Cowpox virus or Vaccinia virus. 23
Traditional laboratory detection of PaPVs includes negative stain transmission electron microscopy (TEM) on skin or scab samples from infected animals, which can clearly distinguish PaPV from other closely related poxviruses such as OPV.2,11 However, TEM facilities are not universally available in veterinary diagnostic laboratories given the high costs of operation. PaPVs may be isolated in cell cultures, but results are not consistent. 17
Several polymerase chain reaction (PCR)–based assays have been reported for rapid detection of PaPVs.10,14,19,25,28 However, these assays were not fully evaluated against elements of performance such as reproducibility, operator variability, and ruggedness (performance of the assay between laboratories), which are critical to ensure accuracy, precision, and fitness for purpose of the assay.15,27
We report herein the development, optimization, and full validation of a highly sensitive TaqMan-based real-time PCR assay for rapid detection of PaPVs. The new assay, referred to as the RVSS (Reagents and Vaccine Services Section) assay was highly specific for PaPV detection only, with no cross-reactivity against other poxviruses. The assay was multiplexed using beta-actin as an internal positive control (IPC), which allowed detection of PCR inhibitors in the clinical specimens and identification of false-negative results.
Materials and methods
Clinical specimens and virus strains
We used 77 PaPV-infected clinical specimens in the form of skin and/or scabs of naturally infected sheep, goats, and cattle, which were collected from different geographical regions of the United States between 2003 and 2005. All specimens were submitted to the USDA Diagnostic Services Section, Foreign Animal Diagnostic Laboratory, and National Veterinary Services Laboratories location in Greenport, NY (hereafter, FADDL) for ruling out foot-and-mouth disease (FMD), which shares clinical signs similar to parapox. Most negative tissues (skin and/or scabs) used in the diagnostic specificity (DSp) study were collected from normal healthy sheep, goats, and cattle from slaughterhouses in the New England area. Some of the negative tissues were kindly provided by the University of Georgia, Athens Veterinary Diagnostic Laboratory (Athens, Georgia). The analytical specificity of the RVSS assay was tested against other viruses, including Foot-and-mouth disease virus (FMDV; serotypes Asia and O), capripoxviruses (Sheeppox virus, Goatpox virus, and Lumpy skin disease virus), Swinepox virus, Raccoonpox virus, and Vaccinia virus (Orthopoxvirus), all of which were obtained from the RVSS except Racconpox and Vaccinia viruses, which were obtained from Cornell University. a
Sample preparation and DNA extraction
Tissue samples were washed in 1× phosphate-buffered saline (PBS) b and suspended in Dulbecco modified Eagle medium c at 10% (w/v) final concentration and homogenized in grinding jars using a lysing agent d for 2 min at an oscillation frequency of 22 Hz. Two hundred microliters of the sample (homogenate or cell culture) were used as starting material for extraction of DNA e ; the extracted DNA was eluted with 100 μL of the kit-supplied elution buffer according to the manufacturer’s instructions.
Transmission electron microscopy
Crude tissue homogenates were clarified by centrifugation at 120 × g for 10 min. The clear supernatant was inoculated onto monolayers of lamb kidney (LK) and/or pig kidney (IBRS-2) cell lines a in Eagle minimum essential medium f with 4% fetal bovine serum. g The growth of the virus was monitored daily based on cytopathic effect following incubation at 37°C in the presence of 5% CO2. The flask was frozen at −70°C and then thawed and centrifuged at 152,000 × g for 30 min using an air-driven ultracentrifuge. h The pellet containing the virus particles was resuspended in 20 μL of sterile water and then transferred to a carbon-coated side of a copper mesh grid. After being stained with 2% phosphotungstic for 2 min, the sample was examined using TEM.
Real-time PCR assay designs
The new RVSS assay targets the highly conserved DNA polymerase gene of the PaPVs genome. The sequences of the primers i and probe j are shown in Table 1. The TaqMan probe j was labeled with FAM (6-carboxyfluorescein) as the reporter dye at the 5′-end and TAMRA (carboxy tetramethylrhodamine) as the quencher dye at the 3′-end. The assays were performed on a thermocycler k and optimized using the DNA extracted from the clinical specimens of PaPVs. The optimal sensitivity of the assay was obtained using a PCR master mix, l with the reaction mix consisting of 1× kit-supplied master mix, 200 nM each of the forward primer, reverse primer, and probe, and 2.5 μL of template plus nuclease-free water to a final volume of 25 μL. The thermocycling conditions included 1 cycle of 95°C for 10 min, and 45 cycles of 95°C for 15 s, and 60°C for 60 s.
The primers and probe sequences of the real-time PCR assays used in the current study.*
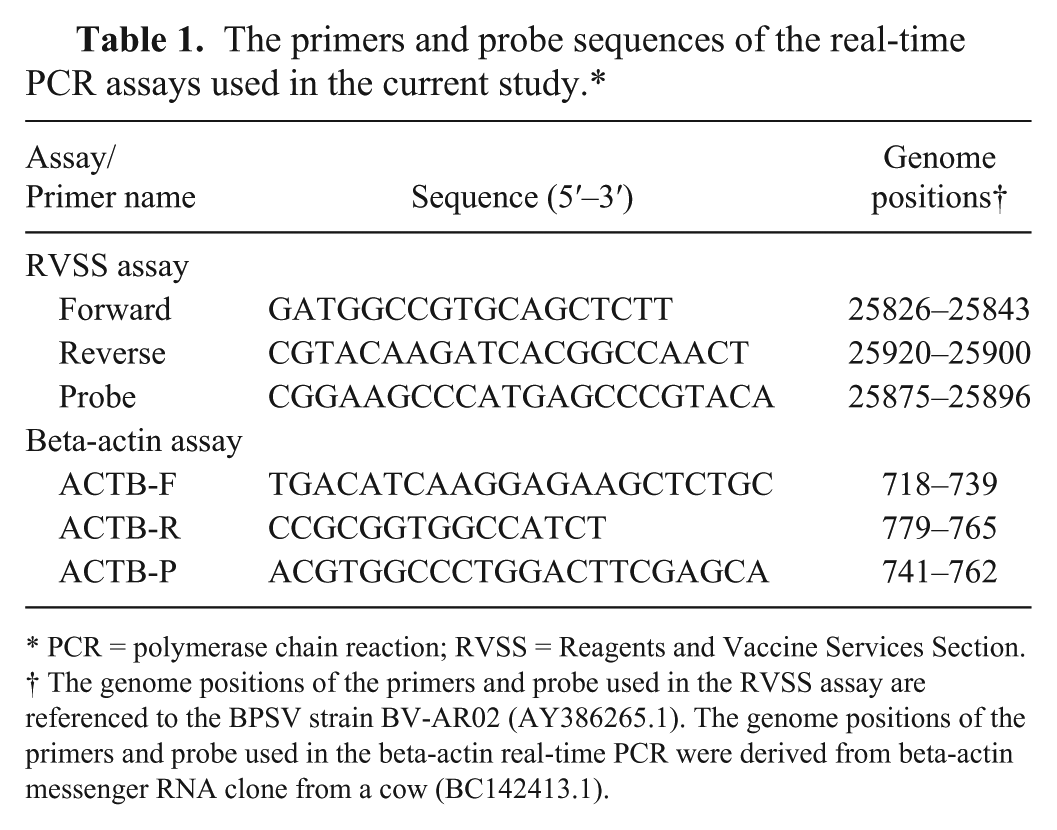
PCR = polymerase chain reaction; RVSS = Reagents and Vaccine Services Section.
The genome positions of the primers and probe used in the RVSS assay are referenced to the BPSV strain BV-AR02 (AY386265.1). The genome positions of the primers and probe used in the beta-actin real-time PCR were derived from beta-actin messenger RNA clone from a cow (BC142413.1).
Five additional published PaPV-specific PCR assays were also used: a generic TaqMan real-time PCR assay targeting the highly conserved major envelope protein gene B2L, 19 referred to as the Nitsche assay; a generic conventional PCR assay, targeting the B2L gene 14 ; and 3 species-specific TaqMan real-time PCR assays (ORFV, BPSV, and PCPV, respectively), each targeting the highly conserved RNA polymerase gene. 28 The Nitsche assay is one of the most commonly used generic PaPV real-time PCR assays in many veterinary diagnostic laboratories in the United States, including FADDL, and therefore we used the latter assay as a reference to compare the performance of the RVSS assay. The species-specific real-time PCR assays 28 were used to determine the species identity of the PaPV isolates in the clinical specimens. The generic conventional PCR assay 14 was used for B2L gene amplification from the clinical specimens followed by DNA sequencing to characterize the PaPV species (described below). The composition of the PCR master mix (25 μL final volume including 2.5 μL of template per assay) and the thermocycling conditions used for the above assays were as described by the authors.14,19,28 Conventional PCR assays were performed on a thermocycler, m and the real-time PCR assays were performed on a different thermocycler. k If not stated otherwise, all assays were performed in replicates, and the average of the data is presented.
Construction of recombinant positive control plasmid
A recombinant positive control plasmid was constructed by amplifying and cloning the PCR amplicon (95 bp) of the RVSS assay into a plasmid vector using a commercial cloning kit. n The recombinant plasmid, referred to as pPPxPC, was transformed into competent Escherichia coli cells o according to the manufacturer’s instructions and purified using a plasmid purification kit. p The authenticity of the PCR clone was further verified by DNA sequencing.
Analytical sensitivity and limit of detection
The analytical sensitivity or the limit of detection (LOD) of the RVSS assay was determined using the DNA extracted from PaPV-infected clinical specimens as template. The PaPV DNA extracted from clinical specimens was 10-fold serially diluted in TE buffer q (10 mM Tris–1 mM EDTA, pH 8.0), and the highest dilution of the virus detectable by the assay was determined as the LOD.
To determine the LOD in terms of copy number, 10-fold serial dilutions of the positive control plasmid pPPxPC in TE q (working stock: 2.67 × 10−5 ng/μL or 6.6 × 105 copies/μL) were used as template. The LOD was expressed as copy number of the plasmid per assay where each copy of the plasmid represents 1 copy of the virus particle. The copy number of the plasmid (pPPxPC) was calculated using the formula: copy number = [mass (g) × 6.022×1023]/[length (bp) × 650 Da].
Intra- and interassay variability
The intra- and interassay variability of the RVSS assay were determined using the DNA extracted from ORFV (sample 616, goat), BPSV (sample 097, cattle), and PCPV (sample CU-8, cattle) as template. Each extracted DNA was appropriately diluted in TE q to obtain high, medium, and low concentrations of the template. The average threshold cycle (Ct) values of the high, medium, and low concentrations of the virus DNA were 19.73, 27.02, and 33.99, respectively, for ORFV; 26.42, 31.08, and 37.12, respectively, for BPSV; and 25.74, 30.34, and 36.64, respectively, for PCPV. Each dilution of the DNA was divided into 5 aliquots, each having sufficient volume to test up to 5 replicates. Five replicates of each dilution (high, medium, or low) were tested from the same aliquot per assay, and the assay was repeated 5 times on different dates. The Ct values corresponding to each dilution (5 × 5 = 25 data points for each dilution) were analyzed to estimate the intra- and interassay variability.
Operator variability
The operator variability of the RVSS assay was determined by an interlaboratory comparison study using the positive control plasmid pPPxPC as template. Ten operators from 5 different veterinary diagnostic laboratories (2 from each laboratory) within the United States, including FADDL, participated in this study. Three different concentrations of the plasmid, strong (Ct: 24.73, SD: 0.69), medium (Ct: 28.51, SD: 0.99), and low (Ct: 31.00, SD: 0.97) were prepared and aliquoted to make multiple panels, each consisting of 6 samples (1 strong, 2 medium, 1 low, and 2 negatives). The operators performing the assay were blinded to the composition of the panel. In addition, each laboratory also received appropriate controls, including PEC (positive extraction control), PAC (positive amplification control), and NEC (negative extraction control). To keep the consistency of the assay at the highest level, each laboratory received the same production lot of the reagents (DNA extraction kit, PCR kit, primers, and probe), and all operators performed the assay on the same type of equipment. k
Diagnostic sensitivity and specificity
Diagnostic sensitivity (DSe) of the RVSS assay was assessed by analyzing 77 PaPV-infected clinical specimens from naturally infected sheep (n = 13), goats (n = 49), cattle (n = 14), and deer (n = 1). The presence of the virus in all specimens was confirmed by negative stain TEM (results not shown). DSp was assessed by analyzing 100 negative tissues (skin and/or scabs) from normal healthy sheep (20), goats (20), and cattle (60). The study was conducted by 5 different veterinary diagnostic laboratories within the United States, including FADDL, and each laboratory received 20 samples (combination of specimens from goat, sheep, and cattle) for testing.
Multiplexing the RVSS assay with beta-actin
Beta-actin is a well-conserved ubiquitous cytoskeletal intracellular protein encoded by the ACTB gene present in all types of clinical specimens. 24 The RVSS assay was multiplexed using beta-actin as IPC. The primers and probe (Table 1) used for amplification of beta-actin were designed from a highly conserved region (64 bp) of the ACTB gene from multiple animal hosts, including cow, sheep, goat, rabbit, swine, and horse (Deng MY, et al. A potentially useful internal positive control for amplification assays on samples of animal origin. Proc 48th AAVLD/USAHA annual meeting, Nov 6–10, 2005, Hershey, PA). The beta-actin TaqMan probe r was labeled with VIC r at the 5′-end as reporter dye and TAMRA at the 3′-end as quencher dye. The composition of the master mix of the multiplex assay included 200 nM each of the forward primer, reverse primer, and probe, added to the singleplex RVSS assay master mix and amplified under the same thermocycling conditions used for the singleplex assay. The amplification of PaPV DNA was monitored in the FAM and that of the beta-actin in the Cy3.
Statistical analysis
All statistical analyses were conducted using R version 3.0.3 (https://www.r-project.org/) and SAS version 9.3 (http://www.sas.com/en_us/home.html). The LOD has been defined as the lowest concentration of analyte at which 95% of samples for that concentration are classified as positive.3,4 The LOD reported in our study is the lowest concentration of the viral DNA in the dilution series at which all replicates test positive as the estimate of the LOD, rather than determining the 95% endpoint from a fitted model. 21
Random coefficient models were fit to the Ct values versus log10 dilution for the LOD data using the current assay. As only a single dilution sequence was tested for the Nitsche assay, a simple linear regression model was used. The amplification efficiency (AE) was estimated as 100 × (10(−1/slope) − 1).
Linear mixed-effects models were used to estimate the intra- and interassay variability as well as the between-laboratory and between-operator within a laboratory variability. 21 All variability estimates were reported as SDs.
DNA sequencing and GenBank accessions
Sanger DNA sequencing reactions were carried out to confirm the identity of the PaPV species used in the LOD studies and to identify ORFV in cattle. The target DNA for the sequencing reactions was the 594-bp partial DNA fragment of the highly conserved major envelope protein gene B2L of the PaPV genome amplified by the generic PaPV conventional PCR assay. 14 The PaPV species of 6 clinical specimens were sequenced: 4 from cattle (samples 097, 466, 508, and CU-8) and 2 from goats (samples 616 and 680). The GenBank accessions of the nucleotide sequences are as follows: KU311138 for sample ID 508, and KU311140–KU311144 for samples 466, 097, CU8, 616, and 680, respectively.
Results
Analytical sensitivity and specificity
The analytical sensitivity (ASe) or the LOD was determined by analyzing serial dilutions of the viral DNA in TE q extracted from BPSV (sample 097), ORFV (sample 616), and PCPV (sample CU-8). For performance evaluation, the LOD was also determined by the Nitsche assay. The LODs for BPSV, ORFV, and PCPV were 10−3, 10−3, and 10−3, respectively, determined by the RVSS assay; and 10−2, 10−3, and 10−3, respectively, determined by the Nitsche assay (Table 2). A subjective cutoff Ct for the RVSS assay was estimated based on the endpoint detection of the virus from serial dilutions using a standard curve. 6 The template DNA was extracted from sample 462 (goat) and 10-fold serially diluted (100–108) in TE. q Based on the Ct values corresponding to the lower LOD of the virus (6 replicates from 2 independent experiments), a cutoff was estimated at 39.09 (Supplemental Fig. 1, available online at http://vdi.sagepub.com/content/by/supplemental-data). This cutoff matched very well with the Ct values of the 77 PaPV-infected clinical specimens that fell within the range 15.20–38.80. Because we did not find any positives beyond 40 cycles of amplification and there were no false positives from either the negative controls (NEC or no-template control NTC) or the negative specimens, we anticipate that the RVSS assay can be performed up to 40 cycles of amplification without loss of sensitivity.
Limit of detection of the RVSS and Nitsche assays.*
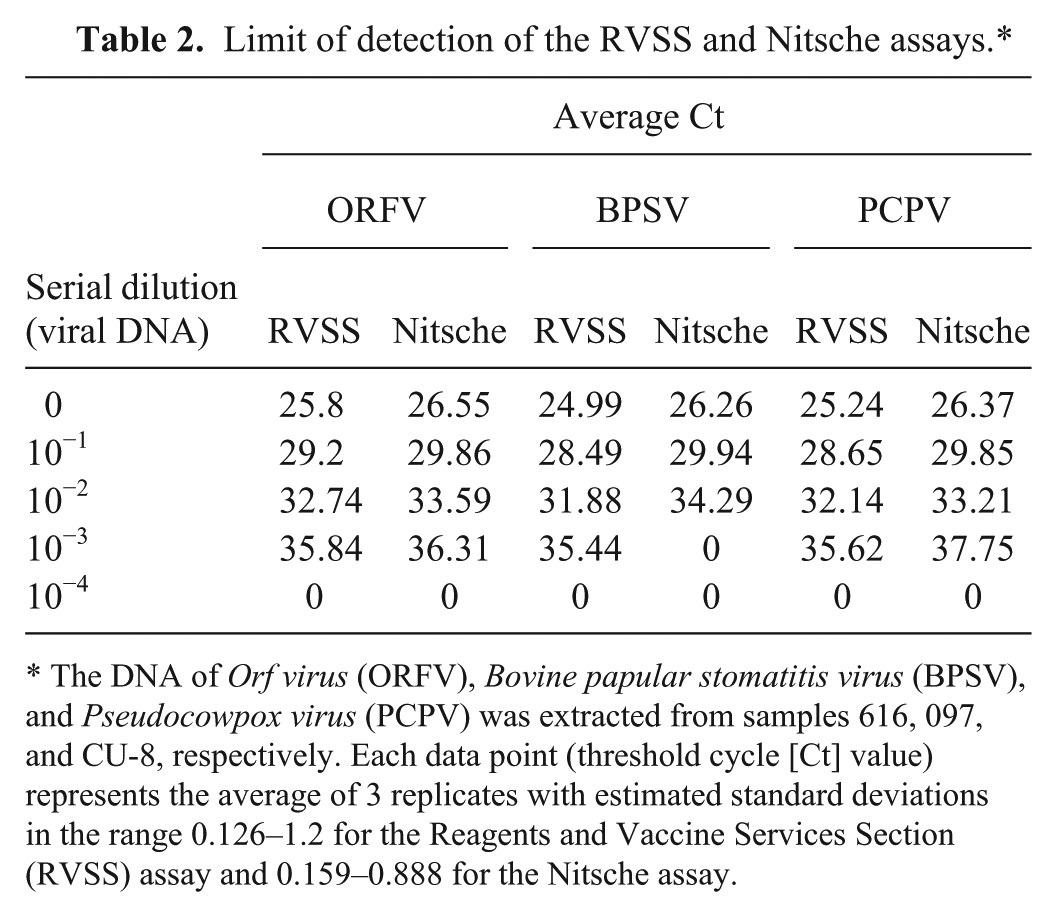
The DNA of Orf virus (ORFV), Bovine papular stomatitis virus (BPSV), and Pseudocowpox virus (PCPV) was extracted from samples 616, 097, and CU-8, respectively. Each data point (threshold cycle [Ct] value) represents the average of 3 replicates with estimated standard deviations in the range 0.126–1.2 for the Reagents and Vaccine Services Section (RVSS) assay and 0.159–0.888 for the Nitsche assay.
The LOD in terms of copy number was determined using serial dilutions of pPPxPC as template. The LOD was determined to be 16 copies per assay, where each copy number of the plasmid represents 1 copy of the virus particle (Fig. 1). The AE (99.1%) and correlation coefficient (R2 = 0.994) estimates calculated from the slope of the standard curve were within the acceptable range and in close agreement with the AE (102.95%) and R2 (0.99) values calculated from serial dilutions of the viral DNA (Supplemental Fig. 1), indicating an approximate doubling of PCR products after each cycle.
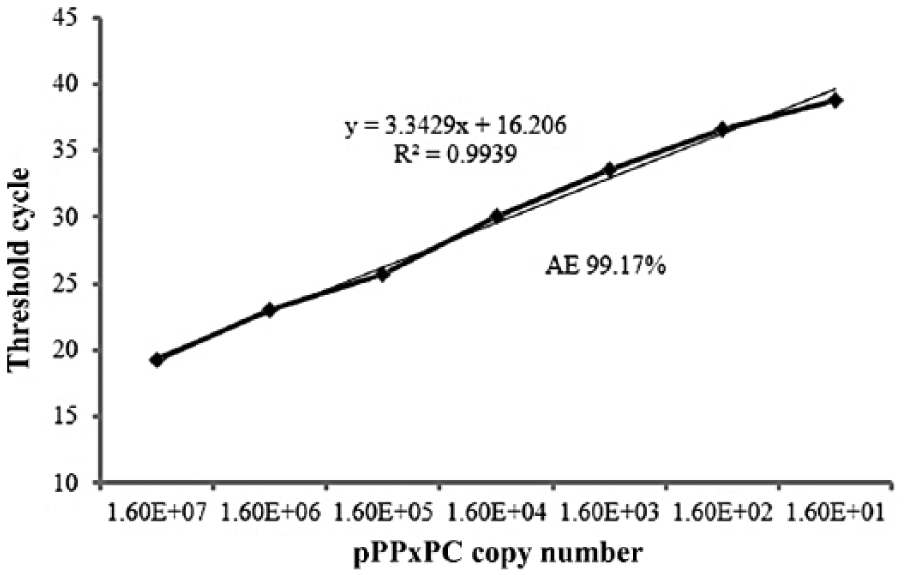
The linear regression standard curve of the Reagents and Vaccine Services Section (RVSS) assay based on the amplification of 10-fold serial dilutions of the positive control plasmid pPPxPC (threshold cycle values vs. the copy number of the plasmid). The regression curve analysis including the slope, correlation coefficient (R2), and amplification efficiency (AE) are shown.
Analytical specificity was determined using multiple clinical specimens of PaPVs from cattle (n = 10), goat (n = 3), and sheep (n = 3), and all tested positive (Ct: 16.38–25.87) by the RVSS assay; there was no cross-reactivity (Ct values zero) found against other viruses including poxviruses (Capripox virus, Swinepox virus, Raccoonpox virus, and Orthopoxvirus) and FMDV.
Diagnostic sensitivity and specificity
All 77 clinical specimens used in the DSe study tested positive for PaPV by RVSS assay (DSe: 100%; Table 4). These specimens were also tested by the Nitsche assay, 19 and 73 tested positive (DSe: 95%; not shown). The 4 specimens that tested negative by the Nitsche assay included 2 from cattle (samples 117, 295), 1 from goat (sample 506), and 1 from deer (sample 750). All 4 specimens had Ct values of 36.56–38.80 determined by the RVSS assay (Table 4). We anticipate the failure to detect the virus in the above specimens could be the result of relatively lower sensitivity of the Nitsche assay. Different assay design and/or PCR master mix might also contribute to the lower DSe of the latter assay.
In the DSp study, all 100 negative specimens from healthy animals tested negative by the RVSS assay with the exception of 1 sample (B6) from a bovid that tested positive for PaPV by one of the participating laboratories, indicating 99% DSp. Further characterization by species-specific real-time PCR indicated the sample was weakly positive (Ct: 39) for BPSV.
Intra- and interassay variability (repeatability)
The intra- and interassay variability estimates of the RVSS assay were calculated based on the Ct values of 3 different concentrations (high, medium, and low) of the DNA extracted from the 3 species of PaPVs (ORFV sample 616, BPSV sample 097, and PCPV sample CU-8) as described in the “Materials and methods” section. For all 3 species of PaPVs, SD estimates of inter-assay variability were <0.01–0.09 and that of the intra-assay variability were 0.20–1.06 (Table 3), which are very low, indicating high reproducibility and repeatability of the RVSS assay.
Inter- and intra-assay variability estimates of the RVSS assay.*
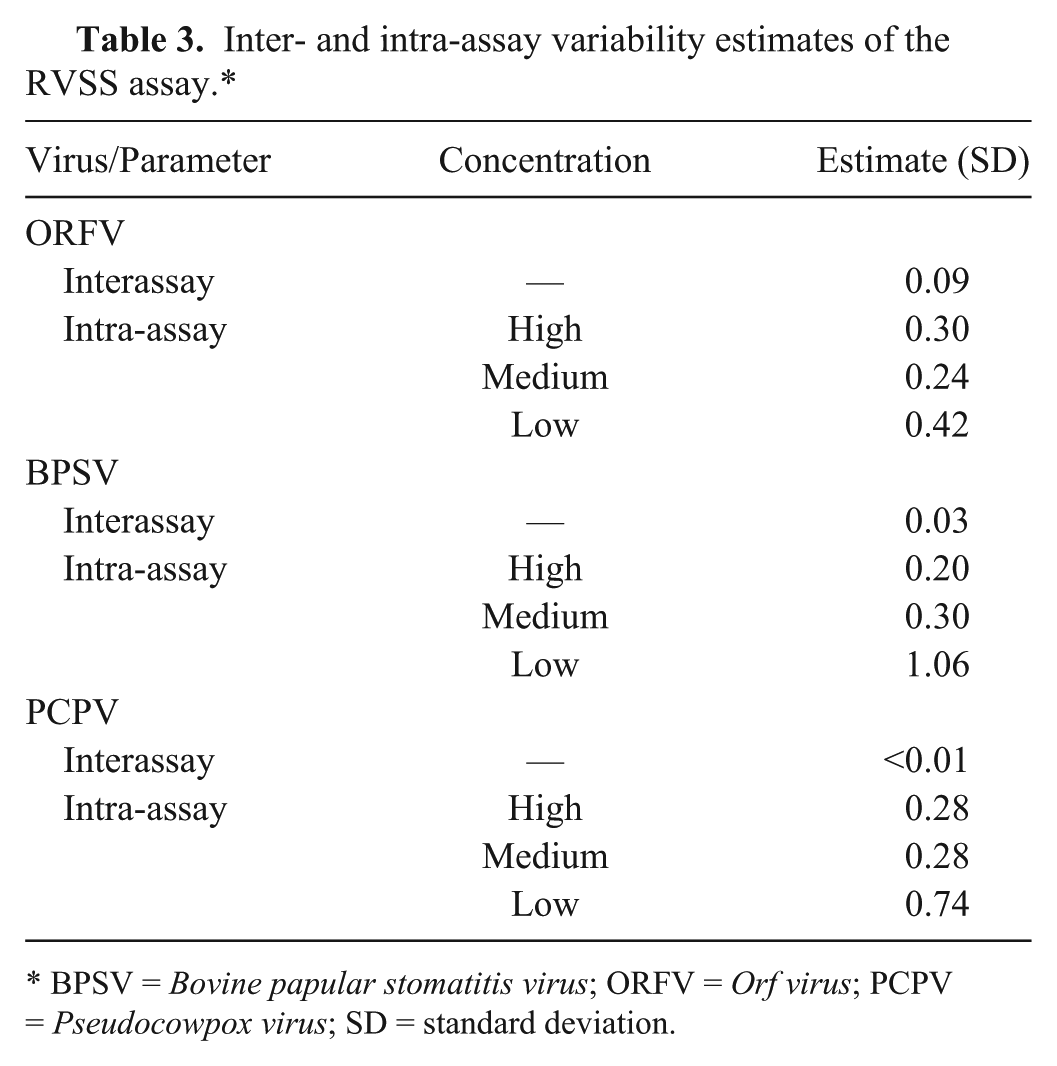
BPSV = Bovine papular stomatitis virus; ORFV = Orf virus; PCPV = Pseudocowpox virus; SD = standard deviation.
Operator variability (reproducibility and ruggedness)
The operator variability of the RVSS assay was assessed using pPPxPC plasmid as described in the “Materials and methods” section. All negative samples including the NEC tested negative, and all positive samples including the PAC and PEC tested positive. The Ct values of the PEC and PAC fell within the acceptable ranges for all operators within each laboratory. Based on the Ct values of the positive samples in the panel, the SD estimates for variability between laboratories and between operators within a laboratory were 0.78 and 0.18, respectively, indicating both reproducibility and tolerance (ruggedness) of the assay against varying test conditions at different laboratories and between operators.
Detection of ORFV in cattle
In 2010, natural infection of ORFV in dairy cows was reported in Brazil. 9 To determine if similar infection(s) occur in the United States, all PaPV-infected clinical specimens (n = 77) used in our study were subjected to ORFV-specific real-time PCR. 28 All specimens from sheep (n = 13) and goats (n = 49) tested positive for ORFV; in addition, 1 specimen from cattle (sample 508) also tested positive for ORFV. Negative stain TEM of the above sample exhibited oval-shaped morphology characteristic for parapoxvirus (Fig. 2). To determine if there was any mixed infection with other PaPVs, the above sample was subjected to BPSV- and PCPV-specific real-time PCR assays, 28 and the results were negative. To characterize the virus, DNA of sample 508 was amplified by generic conventional PCR 14 and sequenced. Analysis of the nucleotide sequence (accession KU31138) by BLAST in the NCBI database revealed higher sequence identities to ORFV (99%) compared to BPSV (85%) and PCPV (94%). For further confirmation, the above sequence was aligned with the corresponding nucleotide sequences of 2 more ORFV specimens from goat (sample 616, KU311143; and sample 680, KU311144) using Kalign (http://msa.sbc.su.se/cgi-bin/msa.cgi), and the alignment revealed 99% identity between themselves, suggesting the presence of ORFV in sample 508.
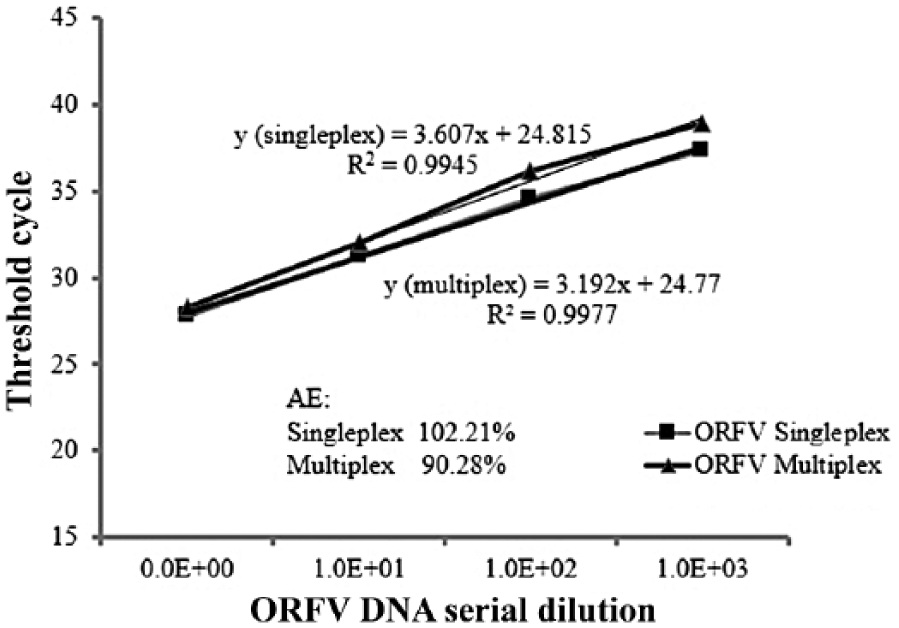
The linear regression standard curve of the Reagents and Vaccine Services Section (RVSS) assay based on the amplification of 10-fold serial dilutions of the DNA extracted from the field isolate Orf virus (ORFV; sample 616, goat) in control blood DNA determined by singleplex (detection of ORFV only) and multiplex (simultaneous detection of ORFV and beta-actin) assays. The regression curve analysis, including the slope, correlation coefficient (R2), and amplification efficiency (AE) are shown.
Multiplexing the RVSS assay with beta-actin
To detect PCR inhibitors (false negatives) in clinical specimens, the RVSS assay was multiplexed using beta-actin as an IPC. To determine if there was any loss of sensitivity resulting from the amplification of beta-actin, both singleplex and multiplex assays were performed using ORFV DNA (sample 616, goat) serially diluted in blood DNA extracted from healthy normal cattle. DNA from blood is used as diluent instead of skin because it is more likely that naturally occurring PCR inhibitors are present in blood than in skin 20 and could be detected using beta-actin as internal control. LOD was determined to be 10−3 dilution for both assays (Fig. 3), indicating no loss of sensitivity because of multiplexing the assay or by PCR inhibitors. AE estimates (90.2–102.2%) and correlation coefficient (R2 = 0.994–0.994) remained within the acceptable range for both singleplex and multiplex assays. Furthermore, beta-actin was detected along with the viral DNA in all dilutions (average Ct: 26.33 ± 0.53).
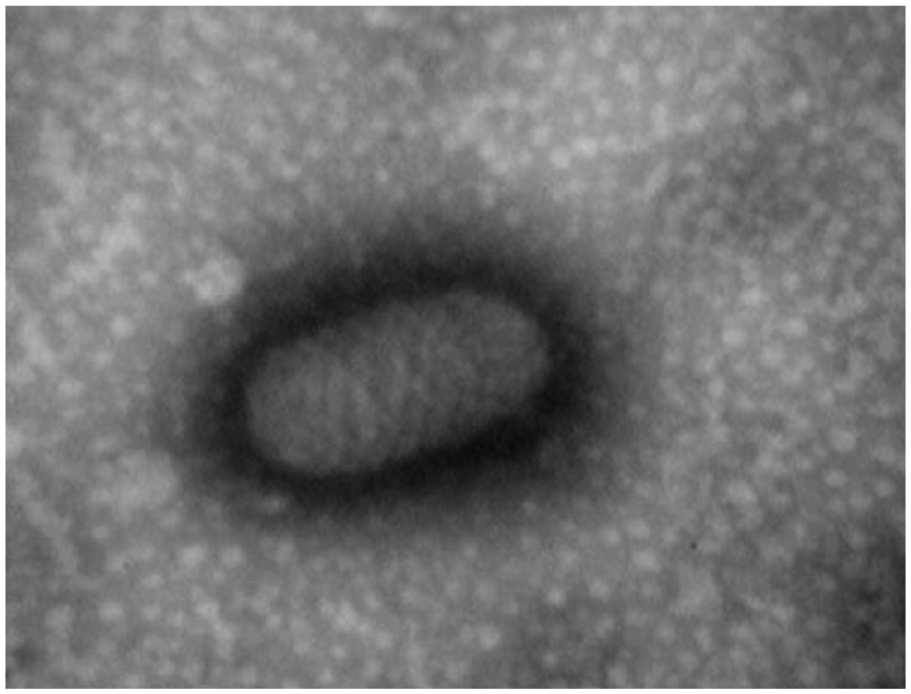
Negative stain transmission electron microscopy of virus particles (200 × 300 nm) of sample 508. 50,000×.
For performance evaluation, the DSe of the multiplex RVSS assay was assessed on 77 PaPV-infected clinical specimens, and all tested positive by both singleplex and multiplex assays along with the detection of beta-actin by the multiplex assay (Table 4), indicating no significant impact on the performance of viral amplifications in the presence of beta-actin–specific primers and probe.
Detection of parapoxvirus (PaPV) and beta-actin in the 77 clinical specimens by singleplex (PaPV only) and multiplex (PaPV plus beta-actin) RVSS assays.*
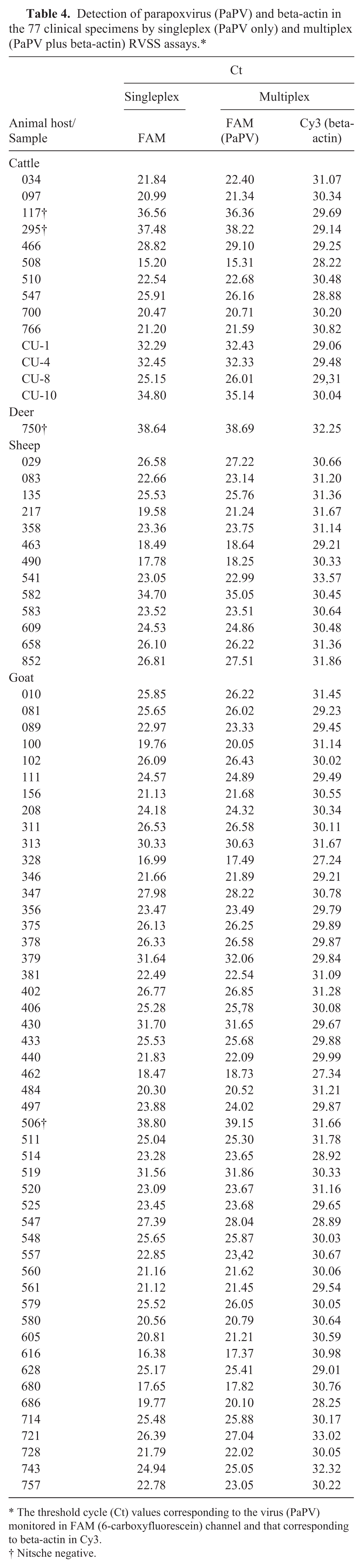
The threshold cycle (Ct) values corresponding to the virus (PaPV) monitored in FAM (6-carboxyfluorescein) channel and that corresponding to beta-actin in Cy3.
Nitsche negative.
Discussion
Parapox is an endemic disease of ruminants in the United States and throughout the world. Although parapox is not an economically important disease by itself, its clinical signs (vesicular and/or skin lesions) closely resemble those of highly contagious vesicular diseases of ruminants such as FMD. 16 Therefore, all PaPV infections are treated as potential FMDV infections until FMDV is ruled out by testing. The clinical signs of PaPV infections in humans also resemble those of orthopoxvirus (such as Cowpox virus or Vaccinia virus) diseases, and cutaneous anthrax or tularemia.2,18 To avoid misdiagnosis, it is essential to have a validated assay in place for accurate and precise detection of parapoxviruses and to differentiate or rule out other look-alike diseases, including FMD, in suspect animals. In this study, we developed and optimized a highly sensitive PaPV real-time PCR assay (RVSS assay) for rapid detection of PaPVs. The RVSS assay was shown to be highly specific for PaPVs (DSe: 100%) and had no cross-reactivity against other poxviruses and FMDV. The new assay (singleplex) was rigorously validated according to the World Organization for Animal Health (OIE) guidelines 27 to meet the quality standards that are critical for veterinary diagnostic laboratories. In addition, the performance of the new assay was evaluated against a published generic PaPV real-time PCR assay (Nitsche assay). 19 Comparison of the analytical and diagnostic sensitivity of the 2 assays indicated slightly higher sensitivity for the RVSS assay compared to the Nitsche assay.
In the DSp study, 99 of 100 specimens from healthy normal animals tested negative (DSp: 99%). The only positive sample (B6) was shown to be weakly positive for BPSV by species-specific real-time PCR. Considering the fact that parapox is endemic in the United States, the occurrence of a single positive specimen in the negative cohort study is not surprising. A 2013 study shows shedding of PaPVs in apparently healthy sheep, goats, and cattle (calves) in livestock markets in the United States. 22
PCR failure (false negative) because of the presence of naturally occurring PCR inhibitors in clinical specimens has been well documented.7,8,12,13,20,26 We multiplexed the RVSS assay using beta-actin as an IPC, which enables the detection of PCR inhibitors and identifies false-negative results. The multiplex assay was shown to simultaneously detect both beta-actin and the virus (PaPVs) without any loss of DSe (Fig. 2, Table 4).
One of the interesting findings in our study was the detection of ORFV as a sole infection in cattle. The presence of ORFV was confirmed by species-specific real-time PCR assays and nucleotide sequencing. In 2013, coinfection of ORFV and Vaccinia virus in cattle was reported. 9 Further study is needed to understand how infections of ORFV occur in an unnatural host (cattle).
Footnotes
Acknowledgements
We thank the following personnel and their institutions for their voluntary participation and contribution in the interlaboratory comparison and negative cohort study: Melissa Laverack and Nancy Zylich (Animal Health Diagnostic Center, College of Veterinary Medicine, Cornell University, Ithaca, New York); Ingrid Fernandez-Marrero and Michelle Norris (Athens Veterinary Diagnostic Laboratory, Athens, Georgia); Beate Crossley, Sonia Mora, and Zaina Iemeir (California Animal Health & Food Safety Laboratory, University of California, Davis, California); Kate Schumann and Karissa Lemire (Diagnostic Services Section, FADDL); and Jianfa Bai and Qing Sun (Veterinary Diagnostic Laboratory, Kansas State University, Manhattan, Kansas). Special thanks to Fredric Grau (Diagnostic Services Section, FADDL) for helping with the Sanger DNA sequencing and analysis.
Authors’ contributions
A Das contributed to conception and design of the study and to acquisition, analysis, and interpretation of data; drafted the manuscript; and critically revised the manuscript. G Ward and A Lowe contributed to conception and design of the study and to acquisition and analysis of data. L Xu contributed to conception and design of the study and to analysis and interpretation of data, and critically revised the manuscript. K Moran contributed to design of the study. R Renshaw and E Dubovi contributed to design of the study and critically revised the manuscript. M Reising contributed to design of the study and to analysis and interpretation of data. W Jia contributed to conception and design of the study; critically revised the manuscript; and gave final approval. All authors agreed to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
a.
RVSS: Reagents and Vaccines Services Section, Foreign Animal Disease Diagnostic Laboratory, Plum Island Animal Disease Center, Greenport, NY.
b.
Life Technologies, Carlsbad, CA.
c.
Life Technologies, Carlsbad, CA.
d.
Qiagen Tissue Lyser, Qiagen Inc., Valencia, CA.
e.
DNeasy Blood and Tissue Kit, Qiagen Inc., Valencia, CA.
f.
Lonza, Anaheim, CA.
g.
Lonza, Anaheim, CA.
h.
Airfuge, Beckman-Coulter Inc., Miami, FL
i.
Integrated DNA Technologies, Coralville, IA.
j.
Life Technologies, Carlsbad, CA.
k.
Cepheid SmartCycler II, Cepheid, Sunnyvale, CA.
l.
Path-ID qPCR Master Mix, Life Technologies, Carlsbad, CA.
m.
Tetrad 2 Peltier Thermocycler, Bio-Rad, Hercules, CA.
n.
TOPO TA Cloning Kit®, Life Technologies, Carlsbad, CA.
o.
Promega Corporation, Madison, WI.
p.
Qiaprep Spin Miniprep plasmid purification kit, Qiagen Inc., Valencia, CA.
q.
Integrated DNA Technologies, Coralville, IA.
r.
Life Technologies, Carlsbad, CA.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the U.S. Department of Agriculture, Animal and Plant Health Inspection Service, National Veterinary Services Laboratories, Ames, Iowa.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.